Document 1
Immunisation Handbook 2026v1 COVID-19 chapter
References
1. V'Kovski P, Kratzel A, Steiner S, et al. Coronavirus biology and replication: implications for SARS-CoV-2.
Nature Reviews: Microbiology, 2021. 19(3): p. 155-170.
2. Wal s AC, Park YJ, Tortorici MA, et al. Structure, function, and antigenicity of the SARS-CoV-2 spike
glycoprotein. Cel , 2020. 181(2): p. 281-292 e6.
3. World Health Organization. 2021 Tracking SARS-CoV-2 variants. WHO; 2021 [updated 13 December
2021]; URL: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/. (accessed 17 December)
4. Ma Q, Liu J, Liu Q, et al. Global percentage of asymptomatic SARS-CoV-2 infections among the tested
population and individuals with confirmed COVID-19 diagnosis: A systematic review and meta-analysis.
JAMA Netw Open, 2021. 4(12): p. e2137257-e2137257.
5. Cevik M, Tate M, Lloyd O, et al. SARS-CoV-2, SARS-CoV, and MERS-CoV viral load dynamics, duration
of viral shedding, and infectiousness: a systematic review and meta-analysis. The Lancet Microbe, 2021.
2(1): p. e13-e22.
6. Bai Y, Yao L, Wei T, et al. Presumed asymptomatic carrier transmission of COVID-19. JAMA, 2020.
323(14): p. 1406-07.
7. Piccoli L, Ferrari P, Piumatti G, et al. Risk assessment and seroprevalence of SARS-CoV-2 infetion in
healthcare workers of COVID-19 and non-COVID-19 hospitals in Southern Switzerland. Lancet Reg Health
Eur, 2021. 1.
8. Liu Y, Yan LM, Wan L, et al. Viral dynamics in mild and severe cases of COVID-19. Lancet Infectious
Diseases, 2020. 20(6): p. 656-7.
9. Lewis N, Chambers LC, Chu HT, et al. Effectiveness associated with vaccination after COVID-19
recovery in preventing reinfection. JAMA Netw Open, 2022. 5(7): p. e2223917-e2223917.
10. Nordstrom P, Bal in M ,Nordstrom A. Risk of SARS-CoV-2 reinfection and COVID-19 hospitalisation in
individuals with natural and hybrid immunity: a retrospective, total population cohort study in Sweden.
Lancet Infectious Diseases, 2022. 22(6): p. 781-790.
11. Powel AA, Dowel AC, Moss P,Ladhani SN. Current state of COVID-19 in children: 4 years on. Journal
of Infection, 2024. 88(5): p. 106134.
12. Aparicio C, Wil is ZI, Nakamura MM, et al. Risk factors for pediatric critical COVID-19: A systematic
review and meta-Analysis. J Pediatric Infect Dis Soc, 2024. 13(7): p. 352-362.
13. Ministry of Health. 2021 Regional Data Explorer 2017-2020: New Zealand Health Survey. URL:
https://www.health.govt.nz/publication/regional-results-2017-2020-new-zealand-health-survey.
(accessed 14 December 2021)
14. Murray S. 2019 The state of wellbeing and equality for disabled people, their families, and whānau.
URL: https://apo.org.au/node/270566. (accessed 14 December 2021)
15. Taylor A, Best EJ, Walls T, et al. COVID-19-related hospitalizations among Aotearoa, New Zealand
children during the Omicron era of SARS-CoV-2. IJID Reg, 2024. 12: p. 100408.
16. Jiang L, Tang K, Levin M, et al. COVID-19 and multisystem inflammatory syndrome in children and
adolescents. Lancet Infectious Diseases, 2020. 20(11): p. e276-e288.
17. Carter MJ, Shankar-Hari M ,Tibby SM. Paediatric inflammatory multisystem syndrome temporal y-
associated with SARS-CoV-2 infection: an overview. Intensive Care Medicine, 2021. 47(1): p. 90-93.
18. Taylor A, Duncanson M, Mitchelson B, et al. Multisystem inflammatory syndrome in New Zealand
children. The Pediatric Infectious Disease Journal, 2023. 42(7): p. e232-e234.
19. Meyerowitz EA, Scott J, Richterman A, et al. Clinical course and management of COVID-19 in the era
of widespread population immunity. Nature Reviews: Microbiology, 2024. 22(2): p. 75-88.
20. Public Health Agency. 2022 COVID-19 mortality in Aotearoa New Zealand: Inequities in risk. .
Wellington. URL: https://www.health.govt.nz/publication/covid-19-mortality-aotearoa-new-zealand-
inequities-risk. (accessed 1 November 2022)
21. Carlson J, Simeone RM, Ellington S, et al. Pre-Delta, Delta, and Omicron periods of the Coronavirus
Disease 2019 (COVID-19) pandemic and health outcomes during delivery hospitalization. Obstetrics and
Gynecology, 2024. 143(1): p. 131-138.
22. Engjom HM, Ramakrishnan R, Vousden N, et al. Severity of maternal SARS-CoV-2 infection and
perinatal outcomes of women admitted to hospital during the omicron variant dominant period using
UK Obstetric Surveillance System data: prospective, national cohort study. BMJ Medicine, 2022. 1(1): p.
e000190.
23. Villar J, Soto Conti CP, Gunier RB, et al. Pregnancy outcomes and vaccine effectiveness during the
period of omicron as the variant of concern, INTERCOVID-2022: a multinational, observational study.
Lancet, 2023. 401(10375): p. 447-457.
24. Al otey J, Stallings E, Bonet M, et al. Clinical manifestations, risk factors, and maternal and perinatal
outcomes of coronavirus disease 2019 in pregnancy: living systematic review and meta-analysis. BMJ,
2020. 370: p. m3320.
25. Mul ins E, Hudak ML, Banerjee J, et al. Pregnancy and neonatal outcomes of COVID-19: co-reporting
of common outcomes from PAN-COVID and AAP SONPM registries. Ultrasound in Obstetrics and
Gynecology, 2021. 57(4): p. 573-581.
26. Munoz FM, Posavad CM, Richardson BA, et al. COVID-19 booster vaccination during pregnancy
enhances maternal binding and neutralizing antibody responses and transplacental antibody transfer to
the newborn. Vaccine, 2023. 41(36): p. 5296-5303.
27. Liguoro I, Pilotto C, Bonanni M, et al. SARS-COV-2 infection in children and newborns: a systematic
review. Eur. J. Pediatrics, 2020. 179(7): p. 1029-1046.
28. Halasa NB, Olson SM, Staat MA, et al. Effectiveness of maternal vaccination with mRNA COVID-19
vaccine during pregnancy against COVID-19–associated hospitalization in infants aged <6 months — 17
States, July 2021–January 2022. MMWR: Morbidity and Mortality Weekly Report, 2022. 71(7): p. 264-
270.
29. Greenhalgh T, Knight M, A'Court C, et al. Management of post-acute covid-19 in primary care. BMJ,
2020. 370: p. m3026.
30. Sivan M ,Taylor S. NICE guideline on long COVID. BMJ, 2020. 371: p. m4938.
31. Sudre CH, Murray B, Varsavsky T, et al. Attributes and predictors of long COVID. Nature Medicine,
2021. 27(4): p. 626-631.
32. Del Rio C, Collins LF ,Malani P. Long-term health consequences of COVID-19. JAMA, 2020. 324(17): p.
1723-1724.
33. Lim JT, Wee LE, Tan JYJ, et al. Multi-systemic risk of post-acute sequelae associated with SARS-CoV-2
reinfection. BMC Glob Public Health, 2025. 3(1): p. 103.
34. Carazo S, Phimmasone J, Skowronski DM, et al. Effectiveness of COVID-19 vaccination and prior
infections to reduce long COVID risk during the pre-Omicron and Omicron periods. Clinical Infectious
Diseases, 2025.
35. World Health Organization. 2023 From emergency response to long-term COVID-19 disease
management: sustaining gains made during the COVID-19 pandemic. WHO; 2023 [updated 3
May 2023]; WHO/WHE/SPP/2023.1:[URL: https://www.who.int/publications/i/item/WHO-WHE-SPP-
2023.1. (accessed February 7)
36. Ioannidis JPA, Pezzullo AM, Cristiano A,Boccia S. Global estimates of lives and life-years saved by
COVID-19 vaccination during 2020-2024. JAMA Health Forum, 2025. 6(7): p. e252223.
37. Geoghegan JL, Ren X, Storey M, et al. Genomic epidemiology reveals transmission patterns and
dynamics of SARS-CoV-2 in Aotearoa New Zealand. Nat Commun, 2020. 11(1): p. 6351.
38. Callaway E. The race for coronavirus vaccines: a graphical guide. Nature, 2020. 580(7805): p. 576-7.
39. Flanagan KL, Best E, Crawford NW, et al. Progress and pitfalls in the quest for effective SARS-CoV-2
(COVID-19) vaccines. Frontiers in Immunology, 2020. 11: p. 579250.
40. Buchan SA, Chung H, Brown KA, et al. Effectiveness of COVID-19 vaccines against Omicron or Delta
symptomatic infection and severe outcomes. medRxiv, 2022 (preprint): p. 2021.12.30.21268565.
41. Mallory RM, Formica N, Pfeiffer S, et al. Safety and immunogenicity following a homologous booster
dose of a SARS-CoV-2 recombinant spike protein vaccine (NVX-CoV2373): a secondary analysis of a
randomised, placebo-control ed, phase 2 trial. Lancet Infectious Diseases, 2022. 22(11): p. 1565-1576.
42. Liu J, Chandrashekar A, Sellers D, et al. Vaccines elicit highly conserved cellular immunity to SARS-
CoV-2 Omicron. Nature, 2022. 603(7901): p. 493-496.
43. Tarke A, Coelho CH, Zhang Z, et al. SARS-CoV-2 vaccination induces immunological T cel memory
able to cross-recognize variants from Alpha to Omicron. Cell, 2022.
44. Andrews N, Tessier E, Stowe J, et al. Duration of protection against mild and severe disease by
COVID-19 vaccines. New England Journal of Medicine, 2022.
45. Happle C, Hoffmann M, Stankov MV, et al. Effects of LP.8.1-adapted mRNA vaccination on SARS-CoV-
2 variant neutralisation. The Lancet Infectious Diseases, 2026. 26(1): p. e3-e5.
46. Polack FP, Thomas SJ, Kitchin N, et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine.
New England Journal of Medicine, 2020. 383(27): p. 2603-2615.
47. Frenck RW, Klein NP, Kitchin N, et al. Safety, immunogenicity, and efficacy of the BNT162b2 COVID-
19 vaccine in adolescents. New England Journal of Medicine, 2021. 385(3): p. 239-250.
48. Walter EB, Talaat KR, Sabharwal C, et al. Evaluation of the BNT162b2 COVID-19 vaccine in children 5
to 11 years of age. New England Journal of Medicine, 2021. 386(1): p. 35-46.
49. Fleming-Dutra KE, Wallace M, Moulia DL, et al. Interim recommendations of the Advisory Committee
on Immunization Practices for use of Moderna and Pfizer-BioNTech COVID-19 vaccines in children aged
6 months–5 years — United States, June 2022. MMWR: Morbidity and Mortality Weekly Report, 2022.
71(26): p. 859-868.
50. Vaccines and Related Biological Products Advisory Committee Meeting. 2022 FDA briefing
document. EUA amendment request for Pfizer-BioNTech COVID-19 vaccine for use in children 6 months
through 4 years of age. URL: https://www.fda.gov/media/159195/download. (accessed 2 February
2023)
51. Dagan N, Barda N, Kepten E, et al. BNT162b2 mRNA Covid-19 vaccine in a nationwide mass
vaccination setting. New England Journal of Medicine, 2021. 384(15): p. 1412-1423.
52. Lopez Bernal J, Andrews N, Gower C, et al. Effectiveness of the Pfizer-BioNTech and Oxford-
AstraZeneca vaccines on covid-19 related symptoms, hospital admissions, and mortality in older adults
in England: test negative case-control study. BMJ, 2021. 373: p. n1088.
53. Birol Ilter P, Prasad S, Berkkan M, et al. Clinical severity of SARS-CoV-2 infection among vaccinated
and unvaccinated pregnancies during the Omicron wave. Ultrasound in Obstetrics and Gynecology,
2022. 59(4): p. 560-562.
54. Intensive Care National Audit and Research Centre (ICNARC). 2022 ICNARC report on COVID-19 in
critical care: England, Wales and Northern Ireland, 8 July 2022. London, UK. URL:
https://www.icnarc.org/our-audit/audits/cmp/reports. (accessed 21 July 2022)
55. Kugelman N, Nahshon C, Shaked-Mishan P, et al. Third trimester messenger RNA COVID-19 booster
vaccination upsurge maternal and neonatal SARS-CoV-2 immunoglobulin G antibody levels at birth.
European Journal of Obstetrics, Gynecology, and Reproductive Biology, 2022. 274: p. 148-154.
56. Rahmati M, Yon DK, Lee SW, et al. Effects of COVID-19 vaccination during pregnancy on SARS-CoV-2
infection and maternal and neonatal outcomes: A systematic review and meta-analysis. Reviews in
Medical Virology, 2023. 33(3): p. e2434.
57. Zerbo O, Ray GT, Fireman B, et al. Maternal SARS-CoV-2 vaccination and infant protection against
SARS-CoV-2 during the first six months of life. Nat Commun, 2023. 14(1): p. 894.
58. Zerbo O, Ray GT, Fireman B, et al. Effectiveness of COVID-19 vaccination during pregnancy by
circulating viral variant. AJOG Glob Rep, 2023. 3(4): p. 100264.
59. Wu S, Li Y, Baral S, et al. Protection of prior SARS-CoV-2 infection, COVID-19 boosters, and hybrid
immunity against Omicron severe illness: A population-based cohort study of five million residents in
Canada. PloS One, 2024. 19(2): p. e0299304.
60. Carreño JM, Alshammary H, Tcheou J, et al. Activity of convalescent and vaccine serum against SARS-
CoV-2 Omicron. Nature, 2021.
61. Lin DY, Xu Y, Gu Y, et al. Effectiveness of bivalent boosters against severe Omicron infection. New
England Journal of Medicine, 2023. 388(8): p. 764-766.
62. Surie D, DeCuir J, Zhu Y, et al. Early estimates of bivalent mRNA vaccine effectiveness in preventing
COVID-19-associated hospitalization among immunocompetent adults aged ≥65 Years - IVY Network, 18
States, September 8-November 30, 2022. MMWR: Morbidity and Mortality Weekly Report, 2022.
71(5152): p. 1625-1630.
63. Huiberts AJ, Hoeve CE, De Gier B, et al. Effectiveness of Omicron XBB.1.5 vaccine against infection
with SARS-CoV-2 Omicron XBB and JN.1 variants, prospective cohort study, the Netherlands, October
2023 to January 2024. Euro Surveil ance, 2024. 29(10).
64. van Werkhoven CH, Valk AW, Smagge B, et al. Early COVID-19 vaccine effectiveness of XBB.1.5
vaccine against hospitalisation and admission to intensive care, the Netherlands, 9 October to 5
December 2023. Euro Surveil ance, 2024. 29(1).
65. Link-Gel es R, Ciesla AA, Mak J, et al. Early estimates of updated 2023-2024 (monovalent XBB.1.5)
COVID-19 vaccine effectiveness against symptomatic SARS-CoV-2 infection attributable to co-circulating
omicron variants among immunocompetent adults - Increasing Community Access to Testing Program,
United States, September 2023-January 2024. MMWR: Morbidity and Mortality Weekly Report, 2024.
73(4): p. 77-83.
66. Wang Q, Guo Y, Bowen A, et al. XBB.1.5 monovalent mRNA vaccine booster elicits robust
neutralizing antibodies against XBB subvariants and JN.1. Cell Host Microbe, 2024. 32(3): p. 315-321 e3.
67. Weidenthaler H, Vidojkovic S, Martin BK,De Moerlooze L. Real-world safety data for MVA-BN:
Increased frequency of syncope fol owing intradermal administration for immunization against mpox
disease. Vaccine, 2024. 42(22): p. 126024.
68. Menni C, Klaser K, May A, et al. Vaccine side-effects and SARS-CoV-2 infection after vaccination in
users of the COVID Symptom Study app in the UK: a prospective observational study. Lancet Infectious
Diseases, 2021. 21(7): p. 939-949.
69. Bobrovitz N, Ware H, Ma X, et al. Protective effectiveness of previous SARS-CoV-2 infection and
hybrid immunity against the omicron variant and severe disease: a systematic review and meta-
regression. Lancet Infectious Diseases, 2023. 23(5): p. 556-567.
70. Blakeway H, Prasad S, Kalafat E, et al. COVID-19 vaccination during pregnancy: coverage and safety.
American Journal of Obstetrics and Gynecology, 2021.
71. Lipkind HS, Vazquez-Benitez G, DeSilva M, et al. Receipt of COVID-19 vaccine during pregnancy and
preterm or small-for-gestational-age at birth - Eight integrated health care organizations, United States,
December 15, 2020-July 22, 2021. MMWR: Morbidity and Mortality Weekly Report, 2022. 71(1): p. 26-
30.
72. Sadarangani M, Soe P, Shulha HP, et al. Safety of COVID-19 vaccines in pregnancy: a Canadian
National Vaccine Safety (CANVAS) network cohort study. Lancet Infectious Diseases, 2022. 22(11): p.
1553-1564.
73. Shimbabukuro T ,CDC-COVID-19 Vaccine Task Force. 2021 COVID-19 vaccine safety update. . (ACIP)
ACoIP. URL: https://www.cdc.gov/vaccines/acip/meetings/slides-2021-1-27-21.html. (accessed 5
February 2021)
74. Faherty EAG, Wilkins KJ, Jones S, et al. Pregnancy outcomes among pregnant persons after COVID-19
vaccination: Assessing vaccine safety in retrospective cohort analysis of U.S. National COVID Cohort
Col aborative (N3C). Vaccines (Basel), 2024. 12(3): p. 289.
75. Perl SH, Uzan-Yulzari A, Klainer H, et al. SARS-CoV-2-specific antibodies in breast milk after COVID-19
vaccination of breastfeeding women. JAMA, 2021. 325(19): p. 2013-2014.
76. Prabhu M, Murphy EA, Sukhu AC, et al. Antibody response to Coronavirus Disease 2019 (COVID-19)
messenger RNA vaccination in pregnant women and transplacental passage into cord blood. Obstetrics
and Gynecology, 2021. 138(2): p. 278-280.
77. Rottenstreich A, Zarbiv G, Oiknine-Djian E, et al. Efficient maternofetal transplacental transfer of
anti- severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike antibodies After antenatal
SARS-CoV-2 BNT162b2 messenger RNA vaccination. Clin Inf Dis, 2021. 73(10): p. 1909-1912.
78. Moro PL, Olson CK, Zhang B, et al. Safety of booster doses of Coronavirus Disease 2019 (COVID-19)
vaccine in pregnancy in the Vaccine Adverse Event Reporting System. Obstetrics and Gynecology, 2022.
140(3): p. 421-427.
79. AusVaxSafety. Pfizer JN.1 COVID-19 vaccine safety data – al participants. COVID-19 vaccines COVID-
19 vaccines; [updated 24 November 2025]; URL: https://www.ausvaxsafety.org.au/vaccine-safety-
data/covid-19-vaccines. (accessed 19 January 2025)
80. Hause AM, Marquez P, Zhang B, et al. Safety monitoring of bivalent COVID-19 mRNA vaccine booster
doses among persons aged ≥12 years - United States, August 31-October 23, 2022. MMWR: Morbidity
and Mortality Weekly Report, 2022. 71(44): p. 1401-1406.
81. Hause AM, Shay DK, Klein NP, et al. Safety of COVID-19 vaccination in United States children ages 5
to 11 years. Pediatrics, 2022. 150(2).
82. Hause AM, Marquez P, Zhang B, et al. COVID-19 mRNA vaccine safety among children aged 6
months-5 years - United States, June 18, 2022-August 21, 2022. MMWR: Morbidity and Mortality
Weekly Report, 2022. 71(35): p. 1115-1120.
83. Edmonds CE, Zuckerman SP ,Conant EF. Management of unilateral axil ary lymphadenopathy
detected on breast MRI in the era of coronavirus disease (COVID-19) vaccination. AJR: American Journal
of Roentgenology, 2021.
84. Garreffa E, Hamad A, O'Sul ivan CC, et al. Regional lymphadenopathy fol owing COVID-19
vaccination: Literature review and considerations for patient management in breast cancer care.
European Journal of Cancer, 2021. 159: p. 38-51.
85. Medsafe. 2022 Adverse events fol owing immunisation with COVID-19 vaccines: Safety Report #40 –
31 January 2022. online. URL: https://www.medsafe.govt.nz/COVID-19/safety-report-40.asp. (accessed
25 February 2022)
86. Pfizer New Zealand. 2021 New Zealand Datasheet: Comirnaty COVID-19 vaccine. Medsafe. URL:
https://www.medsafe.govt.nz/profs/Datasheet/c/comirnatyinj.pdf. (accessed 10 November 2021)
87. Renoud L, Khouri C, Revol B, et al. Association of facial paralysis with mRNA COVID-19 vaccines: A
disproportionality analysis using the World Health Organization pharmacovigilance database. JAMA
Intern Med, 2021. 181(9): p. 1243-1245.
88. Andersson NW, Thiesson EM ,Hvi d A. Adverse events after XBB.1.5-containing COVID-19 mRNA
vaccines. JAMA, 2024. 331(12): p. 1057-1059.
89. Gargano J, Wallace M, Hadler S, et al. Use of mRNA COVID-19 vaccine after reports of myocarditis
among vaccine recipients: Update from Advisory Committee on Immunization Practices - United States,
June 2021. MMWR: Morbidity and Mortality Weekly Report, 2021. 70(27): p. 977-982.
90. Klein N. 2021 Rapid cycle analysis to monitor the safety of COVID-19 vaccines in near real-time
within the Vaccine Safety Datalink: myocarditis and anaphylaxis. Advisory Committee on Immunization
Practices (ACIP). URL: https://www.cdc.gov/vaccines/acip/meetings/downloads/slides-2021-08-30/04-
COVID-Klein-508.pdf. (accessed 20 September 2021)
91. World Health Organization. 2021 COVID-19 subcommittee of the WHO Global Advisory Committee
on Vaccine Safety (GACVS): updated guidance regarding myocarditis and pericarditis reported with
COVID-19 mRNA vaccines.: WHO; 2021 [updated 9 July 2021]; URL: https://www.who.int/news/item/09-
07-2021-gacvs-guidance-myocarditis-pericarditis-covid-19-mrna-vaccines. (accessed 12 July 2021)
92. Diaz GA, Parsons GT, Gering SK, et al. 2021. Myocarditis and pericarditis after vaccination for COVID-
19. JAMA. DOI: 10.1001/jama.2021.13443 (accessed 16 August 2021)
93. Mevorach D, Anis E, Cedar N, et al. Myocarditis after BNT162b2 mRNA vaccine against Covid-19 in
Israel. New England Journal of Medicine, 2021. 385(23): p. 2140-2149.
94. Buchan SA, Seo CY, Johnson C, et al. Epidemiology of myocarditis and pericarditis fol owing mRNA
vaccination by vaccine product, schedule, and interdose interval among adolescents and adults in
Ontario, Canada. JAMA Netw Open, 2022. 5(6): p. e2218505.
95. Shimabukuro TT, Cole M ,Su JR. Reports of anaphylaxis after receipt of mRNA COVID-19 vaccines in
the US-December 14, 2020-January 18, 2021. JAMA, 2021. 325(11): p. 1101-1102.
96. Khalid MB, Zektser E, Chu E, et al. A randomized double-blinded trial to assess recurrence of
systemic allergic reactions following COVID-19 mRNA vaccination. Journal of Al ergy and Clinical
Immunology, 2024. 153(6): p. 1634-1646.
97. Nasreen S, Jiang Y, Lu H, et al. Risk of Guillain-Barre syndrome after COVID-19 vaccination or SARS-
CoV-2 infection: A multinational self-controlled case series study. Vaccine, 2025. 60: p. 127291.
Document 2
NEW ZEALAND DATA SHEET
1. PRODUCT NAME
Comirnaty® LP.8.1 COVID-19 mRNA vaccine, 3 micrograms/0.3 mL dose, concentrate for
suspension for injection (Yellow cap), for age 6 months to 4 years
2. QUALITATIVE AND QUANTITATIVE COMPOSITION
This is a multidose vial that must be diluted before use.
One yellow cap vial (0.48 mL) contains 3 doses of 0.3 mL after dilution, see sections 4.2 and
6.6. One dose (0.3 mL) contains 3 micrograms of SARS-CoV-2 spike protein (mRNA) LP.8.1,
a COVID-19 mRNA Vaccine (embedded in lipid nanoparticles).
SARS-CoV-2 spike protein (mRNA) LP.8.1 is a single-stranded, 5’-capped messenger RNA
(mRNA) produced using a cell-free
in vitro transcription from the corresponding DNA
templates, encoding the viral spike (S) protein of severe acute respiratory syndrome
coronavirus 2 (SARS-CoV-2) (LP.8.1).
For the full list of excipients, see Section 6.1 List of excipients.
3. PHARMACEUTICAL FORM
Concentrate for suspension for injection.
The vaccine is clear to slightly opalescent frozen suspension.
4. CLINICAL PARTICULARS
4.1 Therapeutic indications
Active immunisation to prevent coronavirus disease 2019 (COVID-19) caused by SARS-CoV-
2, in infants and children aged 6 months to 4 years.
The use of this vaccine should be in accordance with official recommendations.
4.2 Dose and method of administration
Dose
Strength & Age Group
Cap and Label Color
Dose Volume
After Dilution
3 micrograms per dose
6 months to 4 years
Yellow
0.3 mL
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 1 of 41
Infants and children 6 months to 4 years of age without history of completion of a
COVID-19 primary course or prior SARS-CoV-2 infection
Comirnaty LP.8.1 3 micrograms/dose is administered intramuscularly after dilution as a
primary course of 3 doses. It is recommended to administer the second dose 3 weeks after the
first dose followed by a third dose administered at least 8 weeks after the second dose (see
sections 4.4 and 5.1).
If a child turns 5 years old between their doses in the primary course, he/she should complete
the primary course at the same 3 micrograms dose level.
Infants and children 6 months to 4 years of age with history of completion of a COVID-
19 primary course or prior SARS-CoV-2 infection
Comirnaty LP.8.1 3 micrograms/dose is administered intramuscularly after dilution as a single
dose for infants and children 6 months to 4 years of age. For individuals who have previously
been vaccinated with a COVID-19 vaccine, Comirnaty LP.8.1 should be administered at least
3 months after the most recent dose of a COVID-19 vaccine.
Comirnaty LP.8.1 (Yellow cap) is for infants and children 6 months to 4 years of age and
cannot be used in individuals 5 years of age and older.
Severely immunocompromised aged 6 months to 4 years
Additional doses may be administered to individuals who are severely immunocompromised
in accordance with national recommendations (see section 4.4).
Interchangeability
The interchangeability of Comirnaty LP.8.1 with other COVID-19 vaccines to complete the
primary vaccination course has not been established. Individuals who have received 1 dose of
Comirnaty LP.8.1 should continue to receive Comirnaty LP.8.1 to complete the primary
vaccination course.
Individuals may not be protected until at least 7 days after their third dose of the vaccine. For
further information on efficacy, see Section 5.1.
Comirnaty LP.8.1 (Yellow cap) should be used only for infants and children 6 months to 4
years of age.
Paediatric population
There are paediatric formulations available for children aged 5 to 11 years of age. For details,
please refer to the data sheets for other fomulations. The safety and efficacy of the vaccine in
infants aged less than 6 months have not yet been established.
Elderly population
Refer to the Data Sheet for Comirnaty LP.8.1suspension for injection (30 micrograms/0.3 mL
dose) (Grey cap) for individuals 12 years of age and older.
Method of administration
Comirnaty LP.8.1 (Yellow cap) should be administered intramuscularly,
after dilution (see
section 6.6).
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 2 of 41
In individuals from 6 to less than 12 months of age, the recommended injection site is the
anterolateral aspect of the thigh. In individuals 1 year of age and older, the recommended
injection site is the anterolateral aspect of the thigh or the deltoid muscle.
Do not inject the vaccine intravascularly, subcutaneously or intradermally.
Comirnaty LP.8.1 should not be mixed in the same syringe with any other vaccines or
medicinal products.
For precautions to be taken before administering the vaccine, see Section 4.4 Special warnings
and precautions for use. For instructions regarding thawing, handling and disposal of the
vaccine, see section 6.6.
Yellow cap vial
After dilution, the yellow cap vials contain 3 doses of 0.3 mL of vaccine.
In order to extract 3 doses from a single vial, low dead-volume syringes and/or needles should
be used. The low dead-volume syringe and needle combination should have a dead volume of
no more than 35 microlitres. If standard syringes and needles are used, there may not be
sufficient volume to extract a tenth dose from a single vial. Irrespective of the type of syringe
and needle:
•
Each dose must contain 0.3 mL of vaccine.
•
If the amount of vaccine remaining in the vial cannot provide a full dose of 0.3 mL, discard
the vial and any excess volume.
•
Do not pool excess vaccine from multiple vials.
For instructions on thawing, handling, dilution and dose preparation of the vaccine before
administration see Section 6.6 Special precautions for disposal and other handling.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of
excipients.
4.4 Special warnings and precautions for use
Traceability
In order to improve the traceability of biological medicinal products, the name and the batch
number of the administered product should be clearly recorded.
General recommendations
Hypersensitivity and anaphylaxis
Events of anaphylaxis have been reported. Appropriate medical treatment and supervision
should always be readily available in case of an anaphylactic reaction following the
administration of Comirnaty.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 3 of 41
The individual should be kept under close observation for at least 15 minutes following
vaccination. A second dose of Comirnaty should not be given to those who have experienced
anaphylaxis to the first dose of Comirnaty.
Myocarditis and pericarditis
Very rare cases of myocarditis and pericarditis have been observed following vaccination with
Comirnaty. These cases have primarily occurred within 14 days following vaccination, more
often after the second vaccination, and more often, but not excluively in younger men. There
have been reports in females. Based on accumulating data, the reporting rates of myocarditis
and pericarditis after primary series in children ages 5 to 11 years are lower than in ages 12 to
17 years. Rates of myocarditis and pericarditis in booster doses do not appear to be higher than
after the second dose in the primary series. The cases are generally mild and individuals tend
to recover within a short time following standard treatment and rest. Cases of myocarditis and
pericarditis following vaccination have rarely been associated with severe outcomes including
death.
Healthcare professionals should be alert to the signs and symptoms of myocarditis and
pericarditis, including atypical presentations. Vaccinees should be instructed to seek
immediate medical attention if they develop symptoms indicative of myocarditis or pericarditis
such as (acute and persisting) chest pain, shortness of breath, or palpitations following
vaccination. Non-specific symptoms of myocarditis and pericarditis also include fatigue,
nausea and vomiting, abdominal pain, dizziness or syncope, oedema and cough. Healthcare
professionals should consult guidance and/or specialists to diagnose and treat this condition.
Stress-related responses
Some individuals may have stress-related responses associated with the process of vaccination
itself. Stress-related responses are temporary and resolve on their own. They may include
dizziness, fainting, palpitations, increases in heart rate, alterations in blood pressure, feeling
short of breath, tingling sensations, sweating and/or anxiety. Individuals should be advised to
bring symptoms to the attention of the vaccination provider for evaluation and precautions
should be in place to avoid injury from fainting.
Concurrent il ness
Vaccination should be postponed in individuals suffering from acute severe febrile illness or
acute infection. The presence of a minor infection and/or low grade fever should not delay
vaccination.
Thrombocytopenia and coagulation disorders
As with other intramuscular injections, Comirnaty LP.8.1 should be given with caution in
individuals receiving anticoagulant ther apy or those with thrombocytopenia or any coagulation
disorder (such as haemophilia) because bleeding or bruising may occur following an
intramuscular administration in these individuals.
Immunocompromised individuals
Immunocompromised persons, including individuals receiving immunosuppressant therapy,
may have a diminished immune response to the vaccine.
Clinical data on safety and immunogenicity after administration of Comirnaty (tozinameran)
in immunocompromised participants are available in 37 participants 2 to 4 years old,
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 4 of 41
65 participants 5 to 11 years old, 15 participants 12 to 17 years old, and 7 participants 18 years
of age and older (see Sections 4.8 Undesirable effects and 5.1 Pharmacodynamic properties).
Duration of protection
The duration of protection afforded by Comirnaty is unknown as it is still being determined by
ongoing clinical trials.
Limitations of vaccine effectiveness
As with any vaccine, vaccination with Comirnaty may not protect all vaccine recipients.
Individuals may not be fully protected until 7 days after their primary course of 3 doses of
Comirnaty.
Use in the elderly
Clinical studies of Comirnaty (tozinameran) include participants 65 years of age and older and
their data contributes to the overall assessment of safety and efficacy. See Section 5.1
Pharmacodynamic properties, Clinical trials, Efficacy against COVID-19.
Paediatric use
The safety and efficacy of Comirnaty in infants aged less than 6 months of age have not yet
been established.
Effects on laboratory tests
No data available.
4.5 Interactions with other medicines and other forms of interactions
No interaction studies have been performed.
Concomitant administration of Comirnaty LP.8.1 (3 micrograms/dose) with other vaccines has
not been studied.
4.6 Fertility, pregnancy and lactation
Comirnaty LP.8.1 (Yellow cap) is not intended for individuals 5 years of age and older.
For details for use in individuals 5 years of age and older, please refer to the Data Sheet for the
relevant strength and presentations.
4.7 Effects on ability to drive and use machines
Comirnaty LP.8.1 has no, or negligible, influence on the ability to drive, cycle and use
machines. However, some of the effects mentioned under Section 4.8 Undesirable effects may
temporarily affect the ability to drive, cycle or use machines.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 5 of 41
4.8 Undesirable effects
Summary of safety profile
The safety of Comirnaty (tozinameran) was evaluated in participants 5 years of age and older
in 3 clinical studies that included 24,675 participants (comprised of 22,026 participants 16
years of age and older, and 1,131 adolescents 12 to 15 years of age from Study C4591001, and
3,109 children 5 to 11 years of age, 2,368 participants 2 to 4 years of age and 1,458 participants
6 to 23 months of age from Study C4591007) that have received at least one dose of Comirnaty
(tozinameran).
Additionally, 306 existing Phase 3 participants at 18 to 55 years of age received a booster dose
of Comirnaty (tozinameran) approximately 6 months after the second dose in the non-placebo-
controlled booster dose portion of Study C4591001. The overall safety profile for the booster
dose was similar to that seen after 2 doses.
In Study C4591031, a placebo-controlled booster study, 5,081 participants 16 years of age and
older were recruited from Study C4591001 to receive a booster dose of Comirnaty
(tozinameran) at least 6 months after the second dose. The overall safety profile for the booster
dose was similar to that seen after 2 doses.
In a subset of C4591007 Phase 2/3 participants, 401 participants 5 to 11 years of age received
a booster dose of Comirnaty at least 5 months after completing the primary series. The overall
safety profile for the booster dose was similar to that seen after the primary series.
In a subset of Study C4591054 (Substudy A, Phase 2/3), 412 participants 12 years of age and
older, who had received at least 3 doses of an mRNA COVID-19 vaccine, received a booster
dose of Comirnaty Omicron XBB.1.5. In another substudy of Study C4591054 (Substudy B,
Phase 2/3), 311 participants 12 years of age and older, who were COVID-19 vaccine-naïve,
received 1 dose of Comirnaty Omicron XBB.1.5. The safety profile of Comirnaty Omicron
XBB.1.5 was similar to the overall Comirnaty safety profile.
In a subset of C4591048 (Substudy E, Phase 2/3), 310 participants 5 to 11 years of age who
were COVID-19 vaccine-naïve, received 1 dose of Comirnaty Omicron XBB.1.5. The safety
profile of Comirnaty Omicron XBB.1.5 was similar to the overall Comirnaty safety profile.
Omicron-adapted Comirnaty
Participants 12 years of age and older – after a single dose in vaccine-naïve individuals
In a subset of C4591054 (Substudy B, Phase 2/3), 311 participants 12 years of age and older,
who were considered to be baseline SARS-CoV-2 positive and COVID-19 vaccine-naïve,
received 1 dose of Comirnaty Omicron XBB.1.5 (raxtozinameran). Participants had a median
follow-up time of 6.4 months up to a data cut-off date of 23 April 2024.
The safety profile of Comirnaty Omicron XBB.1.5 was similar to the overall Comirnaty safety
profile. The most frequent adverse reactions in participants were injection site pain (>50%),
fatigue (>30%), headache (>20%), chills (>10%), diarrhea (>10%), new or worsened muscle
pain (>10%), new or worsened joint pain (>10%), and swelling (>10%).
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 6 of 41
Participants 5 to 11 years of age – after a single dose in vaccine-naïve individuals
In a subset of C4591048 (Substudy E, Phase 2/3), 310 participants 5 to 11 years of age who
were COVID-19 vaccine-naïve, received 1 dose of Comirnaty Omicron XBB.1.5. Participants
had a median follow-up time of 6.4 months up to a data cut-off date of 1 November 2024.
The safety profile of Comirnaty Omicron XBB.1.5 was similar to the overall Comirnaty safety
profile. The most frequent adverse reactions in participants were pain at the injection site
(>40%), fatigue (>10%), headache (>10%), and new or worsened muscle pain (>10%).
Participants 12 years of age and older – after a booster dose
In a subset of C4591054 (Substudy A, Phase 2/3), 412 participants 12 years of age and older,
who had received at least 3 doses of an authorized mRNA COVID-19 vaccine, received a
booster dose of Comirnaty Omicron XBB.1.5.The safety profile of Comirnaty Omicron
XBB.1.5 was similar to the overall Comirnaty safety profile.
COMIRNATY (tozinameran)
Infants 6 to 23 months of age – after 3 doses
In an analysis of Study C4591007 (Phase 2/3), 1,776 infants (1,178 Comirnaty (tozinameran)
3 micrograms and 598 placebo) were 6 to 23 months of age. Based on data in the blinded
placebo-controlled follow-up period up to the cut off date of April 29, 2022, 570 infants 6 to
23 months of age who received a 3 dose primary course [386 Comirnaty (tozinameran) 3
micrograms and 184 placebo] have been followed for a median of 1.3 months after the third
dose.
The most frequent adverse reactions in infants 6 to 23 months of age that received any primary
course dose included irritability (> 60%), decrease appetite (> 30%), tenderness at the injection
site (> 20%), injection site redness and fever (> 10%).
Children 2 to 4 years of age – after 3 doses
In an analysis of Study C4591007 (Phase 2/3), 2,750 children (1,835 Comirnaty (tozinameran)
3 micrograms and 915 placebo) were 2 to 4 years age. Based on data in the blinded placebo-
controlled follow-up period up to the cut off date of April 29, 2022, 886 children 2 to 4 years
of age who received a 3 dose primary course (606 Comirnaty (tozinameran) 3 micrograms and
280 placebo) have been followed a median of 1.4 months after the third dose.
The most frequent adverse reactions in children 2 to 4 years of age that received any primary
course dose included pain at injection site and fatigue (> 40%), injection site redness and fever
(> 10%).
Children 5 to 11 years of age – after 2 doses
In an analysis of Study C4591007 Phase 2/3, 4,647 children (3,109 Comirnaty (tozinameran)
10 micrograms; 1,538 placebo) were 5 to 11 years of age. Of these, 2,206 (1,481 Comirnaty
(tozinameran) 10 micrograms and 725 placebo) children have been followed for >4 months
after the second dose in the placebo-controlled blinded follow-up period. The safety evaluation
in Study C4591007 is ongoing.
The most frequent adverse reactions in children 5 to 11 years of age that received 2 doses
included injection site pain (>80%), fatigue (>50%), headache (>30%), injection site redness
and swelling (≥20%), myalgia, chills and diarrhoea (>10%).
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 7 of 41
Adolescents 12 to 15 years of age – after 2 doses
In an analysis of long term safety follow-up in Study C4591001, 2,260 adolescents
(1,131 Comirnaty (tozinameran) 30 micrograms; 1,129 placebo) were 12 to 15 years of age.
Of these, 1,559 adolescents (786 Comirnaty (tozinameran) and 773 placebo) have been
followed for ≥ 4 months after the second dose of Comirnaty (tozinameran). The safety
evaluation in Study C4591001 is ongoing.
The most frequent adverse reactions in adolescents 12 to 15 years of age that received 2 doses
were injection site pain (>90%), fatigue and headache (>70%), myalgia and chills (>40%),
arthralgia and pyrexia (>20%).
Participants 16 years of age and older – after 2 doses
In Study C4591001, a total of 22,026 participants 16 years of age or older received at least 1
dose of Comirnaty (tozinameran) 30 micrograms and a total of 22,021 participants 16 years of
age or older received placebo (including 138 and 145 adolescents 16 and 17 years of age in the
Comirnaty (tozinameran) and placebo groups, respectively). A total of 20,519 participants 16
years of age or older received 2 doses of Comirnaty (tozinameran).
At the time of the analysis of Study C4591001 with a data cut-off of 13 March 2021 for the
placebo-controlled blinded follow-up period up to the participants’ unblinding dates, a total of
25,651 (58.2%) participants (13,031 Comirnaty (tozinameran) and 12,620 placebo) 16 years of
age and older were followed up for ≥4 months after the second dose. This included a total of
15,111 (7,704 Comirnaty (tozinameran) and 7,407 placebo) participants 16 to 55 years of age
and a total of 10,540 (5,327 Comirnaty (tozinameran) and 5,213 placebo) participants 56 years
and older.
The most frequent adverse reactions in participants 16 years of age and older that received 2
doses were injection site pain (>80%), fatigue (>60%), headache (>50%), myalgia (>40%),
chills (>30%), arthralgia (>20%), pyrexia and injection site swelling (>10%) and were usually
mild or moderate in intensity and resolved within a few days after vaccination. A slightly lower
frequency of reactogenicity events was associated with greater age.
The safety profile in 545 subjects receiving Comirnaty (tozinameran), that were seropositive
for SARS-CoV-2 at baseline, was similar to that seen in the general population.
Study C4591001 also included 200 participants with confirmed stable human
immunodeficiency virus (HIV) infection. The safety profile of the participants receiving
Comirnaty (tozinameran) (n=100) in the individuals with stable HIV infection was similar to
that seen in the general population.
Participants 16 years of age and older – after booster dose
A subset from Study C4591001 Phase 2/3 participants of 306 adults 18 to 55 years of age who
completed the original Comirnaty (tozinameran) 2-dose course, received a booster dose of
Comirnaty (tozinameran) approximately 6 months (range of 4.8 to 8.0 months) after receiving
Dose 2. Of these, 301 participants have been followed for ≥4 months after the booster dose of
Comirnaty (tozinameran).
The most frequent adverse reactions in participants 18 to 55 years of age were injection site
pain (>80%), fatigue (>60%), headache (>40%), myalgia (>30%), chills and arthralgia (>20%).
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 8 of 41
In Study C4591031, a placebo-controlled booster study, participants 16 years of age and older
recruited from Study C4591001 received a booster dose of Comirnaty (tozinameran) (5,081
participants), or placebo (5,044 participants) at least 6 months after the second dose of
Comirnaty (tozinameran). Overall, participants who received a booster dose, had a median
follow-up time of 2.8 months (range 0.3 to 7.5 months) after the booster dose in the blinded
placebo-controlled follow-up period to the cut-off date (8 February 2022). Of these, 1281
participants (895 Comirnaty (tozinameran) and 386 placebo) have been followed for ≥ 4
months after the booster dose of Comirnaty (tozinameran).
Children 5 to 11 years of age – after booster dose
In a subset from C4591007, a total of 401 children 5 to 11 years of age received a booster dose
of Comirnaty (tozinameran) 10 micrograms at least 5 months (range of 5 to 9 months) after
completing the primary series. The analysis of the C4591007 Phase 2/3 subset is based on data
up to the cut-off date of 22 March 2022 (median follow-up time of 1.3 months).
The most frequent adverse reactions in participants 5 to 11 years of age were injection site pain
(>70%), fatigue (>40%), headache (>30%), myalgia, chills, injection site redness, and swelling
(>10%).
Tabulated list of adverse reactions from clinical studies and post-authorisation
experience
Adverse reactions observed during clinical studies are listed below according to the following
frequency categories:
Very common (≥ 1/10),
Common (≥ 1/100 to < 1/10),
Uncommon (≥ 1/1,000 to < 1/100),
Rare (≥ 1/10,000 to < 1/1,000),
Very rare (< 1/10,000),
Not known (cannot be estimated from the available data).
Table 1: Adverse reactions from Comirnaty (tozinameran) and Comirnaty Omicron
XBB.1.5 (raxtozinameran) clinical trials: Individuals 12 years of age and older
Rare
Not known
System Organ
Very common
Common
Uncommon (≥ 1/10,000
(cannot be
Class
(≥ 1/10)
(≥ 1/100 to (≥ 1/1,000 to
estimated from
< 1/10)
< 1/100)
to
< 1/1,000)
the available
data)
Blood and
Lymphadeno
lymphatic system
pathya
disorders
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 9 of 41
Metabolism and
Decreased
nutrition disorders
appetite
Psychiatric
Insomnia
disorders
Nervous system
Headache
Lethargy
Acute
disorders
peripheral
facial
paralysisb
Gastrointestinal
Nausea;
disorders
Skin and
Hyperhidrosi
subcutaneous
s; Night
tissue disorders
sweats
Musculoskeletal
Arthralgia;
and connective
Myalgia
tissue disorders
General disorders Injection site
Injection site Asthenia;
Facial swellingd
and
pain; Fatigue;
redness
Malaise;
administration
Chills; Pyrexiac;
site conditions
Injection site
swelling
a A higher frequency of lymphadenopathy (2.8% vs 0.4%) was observed in participants receiving a booster dose
in Study C4591031 compared to participants receiving 2 doses.
b Through the clinical trial safety follow-up period to 14 November 2020, acute peripheral facial paralysis (or
palsy) was reported by four participants in the Comirnaty (tozinameran) group. Onset was Day 37 after Dose 1
(participant did not receive Dose 2) and Days 3, 9, and 48 after Dose 2. No cases of acute peripheral facial paralysis
(or palsy) were reported in the placebo group.
c A higher frequency of pyrexia was observed after the second dose compared to the first dose. The preferred term
pyrexia is a cluster term covering also body temperature increased.
d Facial swelling in vaccine recipients with a history of injection of dermatological fillers
Table 2. Adverse Reactions from Comirnaty (tozinameran) and Comirnaty Omicron XBB.1.5
(raxtozinameran) clinical trials: Individuals 5 to 11 Years of Age (C4591007 22 May 2022
Data Cut-off Date, C4591048 SSE 10 October 2024 Study Completion Date)
Common Uncommon
Rare
Not known
(cannot be
System Organ
Very
≥1/100 to ≥1/1,000 to ≥1/10,000 to Very Rare estimated
Class
Common
<1/10
<1/100
<1/1,000 <1/10,000
≥1/10 (≥10%) (≥1% to
(≥0.1% to (≥0.01% to (<0.01%)
from the
<10%)
<1%)
<0.1%)
available
data)
Blood and
Lymphadeno
lymphatic system
pathya
disorders
Immune system
Urticariab, c; Angioedema
Anaphylaxisa
disorders
Pruritusb, c; b,c
Rashb, c
Metabolism and
Decreased
nutrition disorders
appetite
Nervous system
Headache
disorders
Gastrointestinal
Diarrhoeab
Vomitingb Nausea
disorders
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 10 of 41
Skin and
Night sweats
subcutaneous tissue
disorders
Musculoskeletal
Myalgia
Arthralgia Pain in
and connective
extremity
tissue disorders
(arm) b
General disorders Injection site Pyrexia
Malaise
and administration pain;
site conditions
Fatigue;
Chills;
Injection site
swelling;
Injection site
redness
a. A higher frequency of lymphadenopathy was observed in C4591007 (1.9% vs. 0.7%) in participants receiving a
booster dose compared to participants receiving 2 doses.
b. These adverse reactions were identified in the post-authorisation period. The following events were not reported
in participants 5 to 11 Years of Age in Study C4591007 but were reported in individuals ≥16 years of age in
Study C4591001: angioedema, lethargy, asthenia, hyperhidrosis, and night sweats.
c. The following events are categorised as hypersensitivity reactions: urticaria, pruritus, rash and angioedema
Table 3. Adverse Reactions from Comirnaty (tozinameran) clinical trial: Individuals 2 to
4 Years of Age (29 April 2022 Data Cut-off Date)
Common Uncommon
Rare
Not known
(cannot be
System Organ Very Common ≥1/100 to ≥1/1,000 to ≥1/10,000 Very Rare estimated
Class
≥1/10
<1/10
<1/100
to <1/1,000 <1/10,000
(≥10%)
(≥1% to
(≥0.1% to (≥0.01% to (<0.01%)
from the
<10%)
<1%)
<0.1%)
available
data)
Blood and
Lymphadeno
lymphatic system
pathy
disorders
Immune system
Rasha,b;
Anaphylaxisa
disorders
Urticariaa,b
Metabolism and
Decreased
nutrition disorders
appetite
Nervous system
Headache
disorders
Gastrointestinal
Diarrhoeaa
Vomitinga
Nausea
disorders
Musculoskeletal and
Myalgia
Pain in
connective tissue
Arthralgia
extremity
disorders
(arm)a
General disorders Injection site
Injection site Asthenia
and administration pain;
swelling;
site conditions
Fatigue;
C hills
Injection site
redness; Pyrexia
* CIOMS frequency categories are based on clinical trial C4591007 crude incidence and was reported to only one
significant figure.
a. These adverse reactions were identified in the post-authorisation period. At the time of the data-lock, the following
reactions were not reported in participants 2 to <5 Years of Age in Study C4591007: pruritus, angioedema, lethargy,
myocarditis, pericarditis, hyperhidrosis, night sweats, and malaise.
b. The following events are categorised as hypersensitivity reactions: rash and urticaria
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 11 of 41
Table 4. Adverse Reactions from Comirnaty (tozinameran) clinical trial: Individuals 6 Months
to 23 months of Age (29 April 2022 Data Cut-off Date)
Not known
Common
Rare
(cannot be
≥1/100 to
Uncommon
≥1/10,000
estimated
Very Common
<1/10
≥1/1,000 to
to <1/1,000 Very Rare from the
System Organ
≥1/10
(≥1% to
<1/100
(≥0.01% to <1/10,000 available
Class
(≥10%)
<10%)
(≥0.1% to <1%) <0.1%) (<0.01%)
data)
Blood and
Lymphadenopathy
lymphatic system
disorders
Immune system
Rasha,b
U
rticariaa,b;
Anaphylaxisa
disorders
Metabolism and
Decreased
nutrition disorders appetite
Psychiatric
Irritability
disorders
Nervous system
Headache
disorders
Lethargy
Gastrointestinal
Vomitinga;
disorders
Diarrhoeaa
General disorders Injection site
Injection Fatigue;
and administration tenderness;
site
Chills
site conditions
Injection site
swelling
redness; Pyrexia
* CIOMS frequency categories are based on clinical trial C4591007 crude incidence and was reported to only one
significant figure.
a. These adverse reactions were identified in the post-authorisation period. At the time of data-lock, the following
events were not reported in participants 6 months to <2 Years of Age in Study C4591007: pruritus, angioedema,
nausea, hyperhidrosis, night sweats, myalgia, arthralgia, pain in extremity, malaise, and asthenia.
b. The following events are categorised as hypersensitivity reactions: rash and urticaria
Immunocompromised participants (adults and children)
In study C4591024, 37 participants 2 to 4 years old, 65 participants 5 to 11 years old, 15
participants 12 to 17 years old, and 7 participants 18 years of age and older from 5 different
immunocompromised disease subsets (immunomodulatory therapy, solid organ transplant,
stem cell transplant, non-small cell lung cancer (NSCLC)/chronic lymphocytic leukaemia
(CLL) and haemodialysis) received at least 1 and up to 4 doses of Comirnaty (tozinameran)
(Doses 1 and 2 were separated by 21 days, Doses 2 and 3 were separated by 28 days and Dose
4 was administered 3 to 6 months after Dose 3).
The safety profile in immunocompromised participants 2 years of age and older who received
Comirnaty (tozinameran) was similar to that in non-immunocompromised participants in other
clinical studies, with no newly identified adverse reactions.
Post-marketing experience
Although the events listed in Table 5 were not observed in the clinical trials, they are considered
adverse drug reactions for Comirnaty as they were reported in the post-marketing experience.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 12 of 41
As these reactions were derived from spontaneous reports, the frequencies could not be
determined and are thus considered as not known.
Table 5: Adverse reactions from Comirnaty post marketing experience
System Organ Class
Adverse Drug Reaction
Immune system disorders
Anaphylaxis
Hypersensitivity reactions (e.g. rash, pruritis, urticaria, angioedema)
Cardiac disorders
Myocarditis
Pericarditis
Nervous system disorders
Dizziness
Gastrointestinal disorders
Diarrhoea
Vomiting
Musculoskeletal and connective Pain in extremity (arm)a
tissue disorders
General disorders and
Extensive swelling of vaccinated limb
administration site conditions
Reproductive system and breast Heavy menstrual bleedingb
disorders
a A higher frequency of pain in extremity (1.1% vs. 0.8%) was observed in participants receiving a booster dose in
Study C4591031 compared to participants receiving 2 doses.
b Most cases appear to be non-serious and temporary in nature.
Reporting suspected adverse effects
Reporting suspected adverse reactions after authorisation of the medicine is important. It allows
continued monitoring of the benefit/risk balance of the medicine. Healthcare professionals are
asked to report any suspected adverse reactions at
https://pophealth.my.site.com/carmreportnz/s/.
4.9 Overdose
In clinical trials, participants who received up to 2 times the recommended dose of Comirnaty
did not have an increase in reactogenicity or adverse reactions.
In post-authorisation experience, there have been reports of higher than recommended doses
of Comirnaty. In general, adverse events reported with overdoses have been similar to the
known adverse reaction profile of Comirnaty.
In the event of overdose, monitoring of vital functions and individualised symptomatic
treatment is recommended.
For risk assessment and advice on the management of overdose please contact the National
Poisons Centre on 0800 POISON (0800 764766).
5. PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: vaccines, other viral vaccines, ATC code: J07BN01.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 13 of 41
Mechanism of action
The nucleoside-modified messenger RNA in Comirnaty is formulated in lipid nanoparticles,
which enable delivery of the non-replicating RNA into host cells to direct transient expression
of the SARS-CoV-2 spike (S) antigen. The mRNA codes for membrane-anchored, full-length
S with two point mutations within the central helix. Mutation of these two amino acids to
proline locks S in an antigenically preferred prefusion conformation. Comirnaty elicits both
neutralising antibody and cellular immune responses to the antigen, which may contribute to
protection against COVID-19.
Clinical efficacy and immunogenicity
Omicron-adapted Comirnaty
Immunogenicity in participants 12 years and older – after a single dose in vaccine-naïve
individuals
In a subset from C4591054, (Substudy B [Phase 2/3]), the evaluable immunogenicity
population of 302 vaccine-naïve participants 12 years of age and older who were considered to
be SARS−CoV-2 positive at baseline, received 1 dose of Comirnaty Omicron XBB.1.5, was
compared with participants in Substudy A [a subset from C4591054, (Phase 2/3)], who
received Comirnaty Omicron XBB.1.5 after at least 3 doses of an mRNA COVID-19 vaccine.
Neutralising titres against Omicron XBB.1.5 increased from baseline to 1 month after study
vaccination and were greater in participants receiving Comirnaty Omicron XBB.1.5 as a single
dose compared with participants who received Comirnaty Omicron XBB.1.5 after at least 3
doses of an mRNA COVID-19 vaccine. Noninferiority was met with respect to the geometric
mean ratio (GMR) of Omicron XBB.1.5-neutralising titres, and the difference in seroresponse
to the XBB.1.5 strain in Substudy B vaccine-naïve participants compared to the subset of
Substudy A (Table 6 and Table 7).
Table 6. Model-Based Geometric Mean Ratio – C4591054 Substudy B and Subset of
Substudy A – Evaluable Immunogenicity Population
Vaccine Group (as Assigned)
Group Comparison
Vaccine-Naïve
Vaccine-Experienced
Substudy B
Substudy A
Comirnaty
Comirnaty
Substudy B /
Omicron XBB.1.5
Omicron XBB.1.5
Substudy A
Sampling
30 mcg
30 mcg
Time
GMTc
GMTc
GMRd
Assaye
Pointa
nb
(95% CIc)
nb
(95% CIc)
(95% CId)
SARS-CoV-2
neutralisation
assay - Omicron
XBB.1.5 - NT50
4373.4
2915.7
1.93
(titre)e
1 month 299 (3757.1, 5090.9) 296
(2462.4, 3452.5)
(1.52, 2.44)f
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre;
LLOQ = lower limit of quantitation; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute respiratory
syndrome coronavirus 2.
a. Protocol-specified timing for blood sample collection.
b. n = Number of participants with valid and determinate assay results for the specified assay at both the
pre−vaccination time point and the given sampling time point.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 14 of 41
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 ×
LLOQ.
d. GMRs and the corresponding 2-sided 95% CIs were calculated by exponentiating the difference in least
square means and the corresponding CIs based on a linear regression model with baseline assay results (log
scale), age, and vaccine group as covariates.
e. SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (Omicron subvariant
XBB.1.5).
f. Noninferiority is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67.
Table 7. Adjusted Difference in Percentages of Participants With Seroresponse –
C4591054 Substudy B and Subset of Substudy A – Evaluable Immunogenicity
Population
Vaccine Group (as Assigned)
Group Comparison
Vaccine-Naïve
Vaccine-Experienced
Substudy B
Substudy A
Comirnaty
Comirnaty
Omicron XBB.1.5
Omicron XBB.1.5
SARS-CoV-2
30 mcg
30 mcg
Adjusted Difference
Neutralisation Sampling
nc (%)
nc (%)
Differenc
Assayg
Time Pointa Nb
(95% CId)
Nb
(95% CId)
e %e
(95% CIf)
SARS-CoV-2
neutralisation
assay - Omicron
XBB.1.5 - NT50
253 (84.9)
218 (73.9)
(titre)g
1 month 298
(80.3, 88.8)
295
(68.5, 78.8)
7.31
(1.34, 13.28)h
Abbreviations: CI = confidence interval; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute respiratory
syndrome coronavirus 2.
a. Protocol-specified timing for blood sample collection.
b. N = number of participants with valid and determinate assay results for the specified assay at both the pre-
vaccination time point and the given sampling time point. These values are the denominators for the
percentage calculations.
c. n = Number of participants with a seroresponse for the given assay at the given sampling time point.
d. Exact 2-sided CI, based on the Clopper and Pearson method.
e. Difference in proportions, expressed as a percentage.
f. 2-Sided CI, based on the Miettinen and Nurminen method stratified by baseline neutralising titre category
(< median, ≥ median) and age group (< median, ≥ median). The median of baseline neutralising titres and
median age was calculated based on the pooled data in 2 comparator groups.
g. SARS-CoV-2 NT50 were determined using a validated 384-well assay platform Omicron subvariant
XBB.1.5).
h. Noninferiority is declared if the lower bound of the 2-sided 95% CI for the difference in percentages of
participants with seroresponse is >-10%.
Immunogenicity in participants 5 to 11 years of age – after a single dose in vaccine-naïve
individuals
In a subset from C4591048 (Substudy E [Phase 2/3]), the evaluable immunogenicity population
of 302 participants, who received a single 10 mcg dose of Comirnaty Omicron XBB.1.5 in
COVID-19 vaccine-naïve participants 5 to 11 years of age was compared to COVID-19
vaccine-experienced participants, 12 to 82 years of age, who received a single 30 mcg dose of
Comirnaty Omicron XBB.1.5 in C4591054 Substudy A. The majority of the participants were
considered to be SARS-CoV-2 positive at baseline (98.9% participants in C4591048 SSE,
99.3% participants in C4591054 Substudy A).
The primary immunobridging analyses compared the geometric mean titres (using a GMR) and
the seroresponse (defined as achieving at least 4-fold rise from baseline) rates in the
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 15 of 41
vaccine−naïve participants 5 to 11 years of age to COVID-19 vaccine-experienced participants
12 years of age and older. The immunobridging criteria were met for both the GMR and the
seroresponse rates (Table 8 and Table 9).
Table 8. Geometric Mean Ratio – C4591048 Substudy E to C4591054 Substudy A -
Participants at 1 Month After the Study Vaccination - Evaluable
Immunogenicity Population
C4591048 SSE
C4591054 SSA
5 to 11 Years of Age
12 Years of Age and older
Comirnaty Omicron XBB.1.5 Comirnaty Omicron XBB.1.5 C4591048 SSE /
SARS-CoV-2
10 mcg
30 mcg
C4591054 SSA
Neutralisation
GMTb
GMTb
GMRc
Assay
na
(95% CIb)
na
(95% CIb)
(95% CIc)
Omicron XBB.1.5
5930.5
4006.4
1.81
- NT50 (titre)d
285
(5283.8, 6656.4)
302
(3438.3, 4668.4)
(1.51, 2.16)e
Abbreviations: CI: confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre; LLOQ =
lower limit of quantitation; LS = least square; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute
respiratory syndrome coronavirus 2; SSA = Substudy A; SSE = Substudy E.
a. n = Number of participants with valid and determinate assay results for the specified assay at the given
sampling time point.
b. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 ×
LLOQ.
c. GMRs and 2-sided 95% CIs were calculated by exponentiating the difference of LS Means for the assay
(C4591048, 5 to 11 years of age – C4591054, 12 years of age and older) and the corresponding CIs based on
a linear regression model with baseline log-transformed neutralising titres, postbaseline infection status, and
vaccine group as covariates.
d. SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (Omicron subvariant
XBB.1.5).
e. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67 and
the point estimate of the GMR is ≥0.8.339
Table 9. Difference in Percentages of Participants With Seroresponse Between
C4591048 Substudy E and C4591054 Substudy A Participants at 1 Month
After the Study Vaccination - Evaluable Immunogenicity Population343
C4591048 SSE
C4591054 SSA
5 to 11 Years of Age
12 Years of Age and older
Comirnaty Omicron XBB.1.5 Comirnaty Omicron XBB.1.5
SARS-CoV-2
10 mcg
30 mcg
Difference
Neutralisation
nb (%)
nb (%)
Assay
Na
(95% CIc)
Na
(95% CIc)
%d
95% CIe
Omicron
XBB.1.5 - NT50
253 (88.8)
231 (77.0)
(3.91,
(titre)f
285
(84.5, 92.2)
300
(71.8, 81.6)
8.97
14.02)g
Abbreviations: CI: confidence interval; LLOQ = lower limit of quantitation; NT50 = 50% neutralising titre;
SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2; SSA = Substudy A; SSE = Substudy E.
Note: Seroresponse is defined as achieving a ≥4-fold rise from baseline. If the baseline measurement is below the
LLOQ, a post-vaccination assay result ≥4 × LLOQ is considered a seroresponse.
a. N = Number of participants with valid and determinate assay results for the specified assay both before
vaccination and at the given sampling time point. This value is the denominator for the percentage calculations.
b. n = Number of participants with seroresponse for the given assay at the given sampling time point.
c. Exact 2-sided 95% CI based on the Clopper and Pearson method.
d. Adjusted difference in proportions based on the Miettinen and Nurminen method stratified by baseline
neutralising titre category (<median, ≥median), expressed as a percentage (C4591048, 5 to 11 years of age –
C4591054, 12 years of age and older). The median of baseline neutralising titres was calculated based on the
pooled data in 2 comparator groups.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 16 of 41
e. 2-sided 95% CI, based on the Miettinen and Nurminen method for the difference in proportions stratified by
baseline neutralising titre category (<median, ≥median), expressed as a percentage.
f. SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (Omicron subvariant
XBB.1.5).
g. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the adjusted difference in percentage
of participants with seroresponse is greater than -10.0%.
Immunogenicity in participants 12 years of age and older – after a booster dose
In a subset from C4591054 (Substudy A, Phase 2/3), the evaluable immunogenicity population
included 382 participants 12 years of age and older who had previously received at least 3 prior
doses of an authorized mRNA COVID-19 vaccine, with the most recent dose being an Omicron
BA.4/BA.5-adapted bivalent vaccine, received a booster dose of Comirnaty Omicron XBB.1.5.
At baseline, 78.8% of participants were considered to be positive for prior SARS−CoV−2
infection.
Compared to participants receiving Comirnaty Original/Omicron BA.4-5 (C4591044),
participants receiving Comirnaty Omicron XBB.1.5 (C4591054) had higher GMTs against
Omicron XBB.1.5 (2622.3 [CI: 2246.6, 3060.9] versus 601.0 [CI: 499.5, 723.1]) and against
Omicron BA.4/BA.5 (5105.1 [CI: 4483.4, 5813.0] versus 4146.0 [CI: 3512.6, 4893.5]) at 1
month after vaccination.
Seroresponse (NT50) was higher against Omicron XBB.1.5, and lower against Omicron
BA.4/BA.5 among participants who received Comirnaty Omicron XBB.1.5 at 1 month
after vaccination compared to the participants who Comirnaty Original/Omicron BA.4-
5 (C4591044) with NT50 against Omicron XBB.1.5 of 73.9% (CI: 69.2%, 78.3%) versus
52.8% (CI: 45.6%, 59.9%), and NT50 against Omicron BA.4/BA.5 of 48.3% (CI: 43.2%,
53.4%) versus 63.0% (CI: 55.9%, 69.7%).Comirnaty (tozinameran)
Efficacy and immunogenicity in individuals 6 months to 4 years of age – 3-dose primary
course
Effectiveness in individuals 6 months to 4 years of age is based on a comparison of efficacy
against symptomatic COVID-19 comparing to placebo and immune responses in this age group
to individuals 16 to 25 years of age.
Efficacy in participants 6 months to 4 years of age – after 3 doses
The efficacy analysis of Study C4591007 was performed across the combined population of
participants 6 months to 4 years of age based on cases confirmed among 873 participants in the
Comirnaty (tozinameran) group and 381 participants in the placebo group (2:1 randomisation
ratio) who received all 3 doses of study intervention during the blinded follow up period when
the Omicron variant of SARS-CoV-2 (BA.2) was the predominant variant in circulation (data
cutoff date of 17 June 2022).
The vaccine efficacy results after Dose 3 in participants 6 months to 4 years of age are
presented in Table 10.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 17 of 41
Table 10: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Dose 3
– Blinded Follow-Up Period – Participants Without Evidence of Infection and
Participants With or Without Evidence of Infection Prior to 7 Days After Dose 3 –
Phase 2/3 – 6 Months to 4 Years of Age – Evaluable Efficacy (3-Dose) Population
First COVID-19 occurrence from 7 days after Dose 3 in participants without evidence of
prior SARS-CoV-2 infection*
Comirnaty
(tozinameran)
3 mcg/Dose
Placebo
Na=873
Na=381
Vaccine Efficacy
Cases n1b
Cases n1b
%
Subgroup
Surveillance Timec (n2d) Surveillance Timec (n2d)
(95% CIe)
13
21
73.2
6 months to 4 yearse
0.124 (794)
0.054 (351)
(43.8, 87.6)
9
13
71.8
2 to 4 years
0.081 (498)
0.033 (204)
(28.6, 89.4)
4
8
75.8
6 months to 23 months
0.042 (296)
0.020 (147)
(9.7, 94.7)
First COVID-19 occurrence from 7 days after Dose 3 in participants with or without evidence
of prior SARS-CoV-2 infection
Comirnaty (tozinameran)
3 mcg/Dose
Placebo
Na=1294
Na=612
Vaccine Efficacy
Cases n1b
Cases n1b
%
Subgroup
Surveillance Timec (n2d) Surveillance Timec (n2d)
(95% CIe)
14
23
72.5
6 months to 4 yearse
0.149 (981)
0.067 (459)
(44.3, 86.9)
10
15
70.7
2 to 4 years
0.100 (639)
0.044 (286)
(30.3, 88.2)
4
8
76.2
6 months to 23 months
0.048 (342)
0.023 (173)
(11.1, 94.8)
Abbreviations: NAAT = nucleic acid amplification test; N-binding = SARS-CoV-2 nucleoprotein–binding;
SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2; VE = vaccine efficacy.
* Participants who had no serological or virological evidence (prior to 7 days after receipt of Dose 3) of past
SARS-CoV-2 infection (i.e., negative N-binding antibody [serum] result at Dose 1, 1 month post-Dose 2
(if available), Dose 3 (if available) visits, SARS-CoV-2 not detected by NAAT [nasal swab] at Dose 1,
Dose 2, and Dose 3 study visits, and a negative NAAT [nasal swab] result at any unscheduled visit prior
to 7 days after receipt of Dose 3) and had no medical history of COVID-19 were included in the analysis.
a. N = number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each
group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 3 to the
end of the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Two-sided 95% confidence interval (CI) for VE is derived based on the Clopper and Pearson method
adjusted for surveillance time.
Analysis of COVID-19 cases that excluded those involving coinfection with other respiratory
pathogens did not meaningfully impact the estimated vaccine efficacy in this population.
Among participants 2 to 4 years of age, severe COVID-19 criteria (as described in the protocol,
based on FDA definition and modified for children) were fulfilled for 9 cases (6 Comirnaty
(tozinameran) and 3 placebo) of which 5 of the 6 cases in the Comirnaty (tozinameran) group
fulfilled a single criterion of increased heart rate or respiratory rate and all 3 cases in the placebo
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 18 of 41
group fulfilled a single criterion of increased heart rate or decreased peripheral oxygen
saturation. None of the cases accrued met criteria for multisystem inflammatory syndrome in
children (MIS-C).
Among participants 6 months to 23 months of age, severe COVID-19 criteria were fulfilled for
3 cases (2 Comirnaty (tozinameran) and 1 placebo) of which 1 of the 2 cases in the Comirnaty
(tozinameran) group fulfilled a single criterion of increased heart rate (152 bpm) and 1 case in
the placebo group fulfilled a single criterion of increased heart rate (172 bpm). None of the
cases accrued met criteria for MIS-C.
Immunogenicity in participants 2 to 4 years of age – after 3 doses
Immunogenicity analyses have been performed in the immunobridging subset of 143
C4591007 participants 2 to 4 years of age without evidence of infection up to 1 month after
Dose 3 based on a data cutoff date of 29 April 2022.
SARS-CoV-2 50% neutralising antibody titres (NT50) were compared between an
immunogenicity subset of Phase 2/3 participants 2 to 4 years of age from C4591007 at 1 month
after the 3-dose primary course and a randomly selected subset from C4591001 Phase 2/3
participants 16 to 25 years of age at 1 month after the 2-dose primary course, using a
microneutralisation assay against the reference strain (USA_WA1/2020). The primary
immunobridging analyses compared the geometric mean titres (using a geometric mean ratio
[GMR]) and the seroresponse (defined as achieving at least 4-fold rise in SARS-CoV-2 NT50
from before Dose 1) rates in the evaluable immunogenicity population of participants without
evidence of prior SARS-CoV-2 infection up to 1 month after Dose 3 in participants 2 to 4 years
of age and up to 1 month after Dose 2 in participants 16 to 25 years of age. The prespecified
immunobridging criteria were met for both the GMR and the seroresponse difference (Table
11 and Table 12, respectively).
Table 11: SARS-CoV-2 GMTs (NT50) at 1 month after vaccination course –
immunobridging subset - participants 2 to 4 years of age (C4591007) 1 month after Dose
3 and participants 16 to 25 years of age (C4591001) 1 month after Dose 2 – without
evidence of SARS-CoV-2 infection – evaluable immunogenicity population
Comirnaty (tozinameran)
3 micrograms/dose
30 micrograms/dose
2 to 4 years of age
16 to 25 years of age
(1 month after Dose 3) (1 month after Dose 2)
GMR (95%CI)
na=143
na=170
(2 to 4 years of age/
Assay
GMTb
GMTb
16 to 25 years of
(95% CIb)
(95% CIb)
age)c,d
SARS-CoV-2
neutralisation assay -
1535.2
1180.0
1.30
NT50 (titre)e
(1388.2, 1697.8)
(1066.6, 1305.4)
(1.13, 1.50)
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre; LLOQ
= lower limit of quantitation; NAAT = nucleic-acid amplification test; NT50 = 50% neutralising titre; SARS-
CoV-2 = severe acute respiratory syndrome coronavirus 2.
Note: Participants who had no serological or virological evidence [(up to 1 month after Dose 2 (C4591001)
or 1 month after Dose 3 (C4591007) blood sample collection)] of past SARS-CoV-2 infection [(i.e., N-
binding antibody [serum] negative at Dose 1, Dose 3 (C4591007) and 1 month after Dose 2 (C4591001) or 1
month after Dose 3 (C4591007), SARS-CoV-2 not detected by NAAT [nasal swab] at Dose 1, Dose 2, and
Dose 3 (C4591007) study visits, and negative NAAT (nasal swab) at any unscheduled visit up to 1 month
after Dose 2 (C4591001) or 1 month after Dose 3 (C4591007) blood collection)] and had no medical history
of COVID-19 were included in the analysis.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 19 of 41
a. n = Number of participants with valid and determinate assay results for the specified assay at the given
dose/sampling time point.
b. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 ×
LLOQ.
c. GMRs and 2-sided 95% CIs were calculated by exponentiating the mean difference of the logarithms of
the titres (2 to 4 years of age minus 16 to 25 years of age) and the corresponding CI (based on the
Student t distribution).
d. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the GMR ratio is greater than
0.67 and the point estimate of the GMR is ≥0.8.
e. SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation
Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus
neutralisation is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum
dilution at which 50% of the virus is neutralised.
Table 12: Difference in percentages of participants with seroresponse at 1 month after
vaccination course – immunobridging subset –participants 2 to 4 years of age (C4591007)
1 month after Dose 3 and participants 16 to 25 years of age (C4591001) 1 month after
Dose 2 without evidence of infection – evaluable immunogenicity population
Comirnaty (tozinameran)
3 micrograms/dose
30 micrograms/dose
2 to 4 years of age
16 to 25 Years of age
Difference in
(1 month after Dose 3) (1 month after Dose 2) seroresponse rates %d
Na=141
Na=170
(95% CIe)
nb (%)
nb (%)
(2 to 4 years of age minus
Assay
(95% CIc)
(95% CIc)
16 to 25 years of age)f
SARS-CoV-2
neutralisation assay -
141 (100.0)
168 (98.8)
NT50 (titre)g
(97.4, 100.0)
(95.8, 99.9)
1.2 (-1.5, 4.2)
Abbreviations: LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test; N-binding
= SARS-CoV-2 nucleoprotein–binding; NT50 = 50% neutralising titre 50; SARS-CoV-2 = severe acute
respiratory syndrome coronavirus 2.
Note: Seroresponse is defined as achieving a ≥4-fold rise from baseline (before Dose 1). If the baseline
measurement is below the LLOQ, a postvaccination assay result ≥4 × LLOQ is considered a seroresponse.
Note: Participants who had no serological or virological evidence (up to 1 month after Dose 2 [(C4591001) or 1
month after Dose 3 (C4591007) blood sample collection)[ of past SARS-CoV-2 infection [(i.e., N-binding
antibody [serum] negative at pre-Dose 1, pre-Dose 3 (C4591007) and 1 month after Dose 2 (C4591001) or 1
month after Dose 3 (C4591007), SARS-CoV-2 not detected by NAAT [nasal swab] at pre-Dose 1, pre-Dose 2,
and pre-Dose 3 (C4591007) study visits, and negative NAAT (nasal swab) at any unscheduled visit up to
1 month after Dose 2 (C4591001) or 1 month after Dose 3 (C4591007) blood collection)] and had no medical
history of COVID-19 were included in the analysis.
a. N = number of participants with valid and determinate assay results both before vaccination and at 1 month
after Dose 2. These values are the denominators for the percentage calculations.
b. n = Number of participants with seroresponse for the given assay at the given dose/sampling time point.
c. Exact 2-sided CI based on the Clopper and Pearson method.
d. Difference in proportions, expressed as a percentage (2 to 4 years of age minus 16 to 25 years of age).
e. 2-sided CI, based on the Miettinen and Nurminen method for the difference in proportions, expressed as a
percentage.
f. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the difference in proportions is
greater than -10.0% provided that the immunobridging criteria based on GMR were met.
g. SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation
Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus
neutralisation is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution
at which 50% of the virus is neutralised.
Using a non-validated fluorescence focus reduction neutralisation test assay against the
Omicron variant of SARS-CoV-2 (BA.1), the NT50 GMT at 1 month after Dose 3 among a
subset of 34 study participants without evidence of prior SARS-CoV-2 infection (82.5 [2-sided
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 20 of 41
95% CI: 55.4, 122.9]) was increased compared to the NT50 GMT before Dose 3 (14.0 [2-sided
95% CI: 10.6, 18.5]).
An additional descriptive immunogenicity analysis was performed for participants 2 to 4 years
of age who received a 3-dose course of Comirnaty (tozinameran) in Phase 2/3 C4591007,
compared with a subset of participants 18 to 50 years of age in Phase 3 Study C4591017 who
had received a 2-dose primary course followed by a booster dose of Comirnaty (tozinameran)
30 micrograms. The comparator group (participants 18 to 50 years of age) in this analysis had
a similar interval between Comirnaty (tozinameran) Dose 2 and Dose 3 (median 13.0 weeks)
as the participants 2 to 4 years of age (median 10.6 weeks). Among 34 participants 2 to 4 years
of age without evidence of prior SARS-CoV-2 infection who received 3 doses of Comirnaty
(tozinameran) 3 micrograms, neutralising GMTs were 114.3 at 1-month post-Dose 3. Among
27 participants 18 to 50 years of age without evidence of prior SARS-CoV-2 infection who
received 3 doses of Comirnaty (tozinameran) 30 micrograms, Omicron neutralising GMTs
were 164.2 at 1-month post Dose 3.
Immunogenicity in participants 6 to 23 months of age – after 3 doses
Immunogenicity analyses have been performed in the immunobridging subset of 82 C4591007
participants 6 months to 23 months of age without evidence of infection up to 1 month after
Dose 3 based on a data cutoff date of 29 April 2022.
SARS-CoV-2 50% neutralising antibody titres (NT50) 1 month after the vaccination course
were compared between an immunogenicity subset of Phase 2/3 participants 6 months to 23
months of age from C4591007 and a randomly selected subset from C4591001 Phase 2/3
participants 16 to 25 years of age, using a microneutralisation assay against the reference strain
(USA_WA1/2020). The primary immunobridging analyses compared the geometric mean
titres (using a GMR) and the seroresponse (defined as achieving at least 4-fold rise in SARS-
CoV-2 NT50 from before Dose 1) rates in the evaluable immunogenicity population of
participants without evidence of prior SARS-CoV-2 infection up to 1 month after Dose 3 in
participants 6 months to 23 months of age and up to 1 month after Dose 2 in participants 16 to
25 years of age. The prespecified immunobridging criteria were met for both the GMR and the
seroresponse difference (Table 13 and Table 14, respectively).
Table 13: SARS-CoV-2 GMTs (NT50) at 1 month after vaccination course –
immunobridging subset - participants 6 months to 23 months of age (C4591007) 1 month
after Dose 3 and participants 16 to 25 years of age (C4591001) 1 month after Dose 2 –
without evidence of SARS-CoV-2– evaluable immunogenicity population
Comirnaty (tozinameran)
3 micrograms/dose
6 months to 23 months
30 micrograms/dose
of age
16 to 25 years of age
(1 month after Dose 3) (1 month after Dose 2)
GMR (95%CI)
na=82
na=170
(6 months to 23 months
Assay
GMTb
GMTb
of age/16 to 25 years of
(95% CIb)
(95% CIb)
age)c,d
SARS-CoV-2
neutralisation assay -
NT50 (titre)e
1406.5 (1211.3, 1633.1) 1180.0 (1066.6, 1305.4)
1.19 (1.00, 1.42)
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre; LLOQ =
lower limit of quantitation; NAAT = nucleic-acid amplification test; NT50 = 50% neutralising titre; SARS-CoV-2
= severe acute respiratory syndrome coronavirus 2.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 21 of 41
Note: Participants who had no serological or virological evidence [(up to 1 month after Dose 2 (C4591001) or 1
month after Dose 3 (C4591007) blood sample collection)] of past SARS-CoV-2 infection (i.e., N-binding
antibody [serum] negative at Dose 1, Dose 3 (C4591007) and 1 month after Dose 2 (C4591001) or 1 month after
Dose 3 (C4591007), SARS-CoV-2 not detected by NAAT [nasal swab] at Dose 1, Dose 2, and Dose 3
(C4591007) study visits, and negative NAAT (nasal swab) at any unscheduled visit up to 1 month after Dose 2
(C4591001) or 1 month after Dose 3 (C4591007) blood collection)] and had no medical history of COVID-19
were included in the analysis.
a. n = Number of participants with valid and determinate assay results for the specified assay at the given
dose/sampling time point.
b. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titre
titres and the corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
c. GMRs and 2-sided 95% CIs were calculated by exponentiating the mean difference of the logarithms of the
titres (6 months to 23 months of age minus 16 to 25 years of age) and the corresponding CI (based on the
Student t distribution).
d. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the GMR ratio is greater than 0.67
and the point estimate of the GMR is ≥0.8.
e. SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation Assay.
The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus neutralisation is
read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution at which 50% of
the virus is neutralised.
Table 14: Difference in percentages of participants with seroresponse at 1 month after
vaccination course – immunobridging subset – participants 6 months to 23 months of age
(C4591007) 1 month after Dose 3 and participants 16 to 25 years of age (C4591001) to
1 month after Dose 2 without evidence of infection – evaluable immunogenicity
population
Comirnaty (tozinameran)
3 micrograms/dose
30 micrograms/dose
6 to 23 months
16 to 25 years
Difference in
of age
of age
seroresponse rates %d
(1 month after Dose 3) (1 month after Dose 2)
(95% CIe)
Na=80
Na=170
(6 months to 23
Assay
nb (%)
nb (%)
months of age minus
(95% CIc)
(95% CIc)
16 to 25 years of age)f
SARS-CoV-2
neutralisation assay -
80 (100.0)
168 (98.8)
NT50 (titre)g
(95.5, 100.0)
(95.8, 99.9)
1.2 (-3.4, 4.2,)
Abbreviations: LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test; N-binding
= SARS-CoV-2 nucleoprotein–binding; NT50 = 50% neutralising titre 50; SARS-CoV-2 = severe acute
respiratory syndrome coronavirus 2.
Note: Seroresponse is defined as achieving a ≥4-fold rise from baseline (before Dose 1). If the baseline
measurement is below the LLOQ, a postvaccination assay result ≥4 × LLOQ is considered a seroresponse.
Note: Participants who had no serological or virological evidence [(up to 1 month after Dose 2 (C4591001) or 1
month after Dose 3 (C4591007) blood sample collection) of past SARS-CoV-2 infection (i.e., N-binding
antibody [serum] negative at pre-Dose 1, Dose 3 (C4591007) and 1 month after Dose 2 (C4591001) or 1 month
after Dose 3 (C4591007), SARS-CoV-2 not detected by NAAT [nasal swab] at pre-Dose 1, pre-Dose 2, and
pre-Dose 3 (C4591007) study visits, and negative NAAT (nasal swab) at any unscheduled visit up to 1 month
after Dose 2 (C4591001) or 1 month after Dose 3 (C4591007) blood collection)] and had no medical history of
COVID-19 were included in the analysis.
a. N = number of participants with valid and determinate assay results both before vaccination and at 1 month
after Dose 2. These values are the denominators for the percentage calculations.
b. n = Number of participants with seroresponse for the given assay at the given dose/sampling time point.
c. Exact 2-sided CI based on the Clopper and Pearson method.
d. Difference in proportions, expressed as a percentage (6 months to 23 months of age minus 16 to 25 years of
age).
e. 2-sided CI, based on the Miettinen and Nurminen method for the difference in proportions, expressed as a
percentage.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 22 of 41
f. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the difference in proportions is
greater than -10.0% provided that the immunobridging criteria based on GMR were met.
g. SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation
Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus
neutralisation is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution
at which 50% of the virus is neutralised.
Using a non-validated fluorescence focus reduction neutralisation test assay against the
Omicron variant of SARS-CoV-2 (BA.1), the NT50 GMT at 1 month after Dose 3 among a
subset of 32 study participants without evidence of prior SARS-CoV-2 infection (127.5 [2-
sided 95% CI: 90.2, 180.1]) was increased compared to the NT50 GMT before Dose 3 (16.3
[2-sided 95% CI: 12.8, 20.8]).
An additional descriptive immunogenicity analysis was performed for participants 6 months to
23 months of age who received a 3-dose course of Comirnaty (tozinameran) in Phase 2/3
C4591007, compared with a subset of participants 18 to 50 years of age in Phase 3 Study
C4591017 who had received a 2-dose primary course followed by a booster dose of Comirnaty
(tozinameran) 30 micrograms. The comparator group (participants 18 to 50 years of age) in
this analysis had a similar interval between Comirnaty (tozinameran) Dose 2 and Dose 3
(median 13.0 weeks) as the participants 6 months to 23 months of age (median 12.9 weeks).
Among 32 participants 6 months to 23 months of age without evidence of prior SARS-CoV-2
infection who received 3 doses of Comirnaty (tozinameran) 3 micrograms, Omicron
neutralising GMTs were 128.8 at 1-month post-Dose 3. Among 27 participants 18 to 50 years
of age without evidence of prior SARS-CoV-2 infection who received 3 doses of Comirnaty
(tozinameran) 30 micrograms, Omicron neutralising GMTs were 164.2 at 1-month post Dose
3.
Efficacy in other age groups
Study C4591001 is a multicentre, multinational, Phase 1/2/3 randomised, placebo-controlled,
observer-blind dose-finding, vaccine candidate selection and efficacy study in participants 12
years of age and older. Randomisation was stratified by age: 12 to 15 years of age, 16 to 55
years of age, or 56 years of age and older, with a minimum of 40% of participants in the ≥56-
year stratum. The study excluded participants who were immunocompromised and those who
had previous clinical or microbiological diagnosis of COVID-19. Participants with pre-existing
stable disease, defined as disease not requiring significant change in therapy or hospitalisation
for worsening disease during the 6 weeks before enrolment, were included as were participants
with known stable infection with HIV, hepatitis C virus (HCV) or hepatitis B virus (HBV).
Efficacy in participants 16 years of age and older – after 2 doses
In the Phase 2/3 portion of Study C4591001, based on data accrued through
14 November 2020, approximately 44,000 participants were randomised equally and were to
receive 2 doses of Comirnaty (tozinameran) or placebo. The efficacy analyses included
participants that received their second vaccination within 19 to 42 days after their first
vaccination. The majority (93.1%) of vaccine recipients received the second dose 19 days to
23 days after Dose 1. Participants are planned to be followed for up to 24 months after Dose 2,
for assessments of safety and efficacy against COVID-19. In the clinical study, participants
were required to observe a minimum interval of 14 days before and after administration of an
influenza vaccine in order to receive either placebo or Comirnaty (tozinameran). In the clinical
study, participants were required to observe a minimum interval of 60 days before or after
receipt of blood/plasma products or immunoglobulins through to conclusion of the study in
order to receive either placebo or Comirnaty (tozinameran).
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 23 of 41
The population for the analysis of the primary efficacy endpoint included 36,621 participants
12 years of age and older (18,242 in the Comirnaty (tozinameran) group and 18,379 in the
placebo group) who did not have evidence of prior infection with SARS-CoV-2 through 7 days
after the second dose. In addition, 134 participants were between the ages of 16 to 17 years of
age (66 in the Comirnaty (tozinameran) group and 68 in the placebo group) and 1616
participants 75 years of age and older (804 in the Comirnaty (tozinameran) group and 812 in
the placebo group).
At the time of the primary efficacy analysis, participants had been followed for symptomatic
COVID-19 for in total 2,214 person-years for the Comirnaty (tozinameran) group and in total
2,222 person-years for the placebo group.
There were no meaningful clinical differences in overall vaccine efficacy in participants who
were at risk of severe COVID-19 including those with 1 or more comorbidities that increase
the risk of severe COVID-19 (e.g. asthma, body mass index (BMI) ≥30 kg/m2, chronic
pulmonary disease, diabetes mellitus, hypertension).
Comirnaty (tozinameran) efficacy information is presented in Table 15.
Table 15: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2, by age
subgroup – participants without evidence of infection prior to 7 days after Dose 2 –
evaluable efficacy (7 days) population
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence of prior
SARS-CoV-2 infection*
Comirnaty
(tozinameran)
Placebo
Vaccine efficacy
Subgroup
Na = 18,198
Na = 18,325
%
Cases n1b
Cases n1b
(95% CI)f
Surveillance timec (n2d)
Surveillance timec (n2d)
All participantse
8
162
95.0
2.214 (17,411)
2.222 (17,511)
(90.0, 97.9)
16 to 64 years
7
143
95.1
1.706 (13,549)
1.710 (13,618)
(89.6, 98.1)
65 years and older
1
19
94.7
0.508 (3848)
0.511 (3880)
(66.7, 99.9)
65 to 74 years
1
14
92.9
0.406 (3074)
0.406 (3095)
(53.1, 99.8)
75 years and older
0
5
100.0
0.102 (774)
0.106 (785)
(-13.1, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction
(RT-PCR) and at least 1 symptom consistent with COVID-19 [*Case definition: (at least 1 of) fever,
new or increased cough, new or increased shortness of breath, chills, new or increased muscle pain,
new loss of taste or smell, sore throat, diarrhoea or vomiting.]
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the last
dose) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and
SARS-CoV-2 not detected by nucleic acid amplification tests (NAAT) [nasal swab] at Visits 1 and
2), and had negative NAAT (nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were
included in the analysis.
a. N = number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within
each group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after
Dose 2 to the end of the surveillance period.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 24 of 41
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence of prior
SARS-CoV-2 infection*
Comirnaty
(tozinameran)
Placebo
Vaccine efficacy
Subgroup
Na = 18,198
Na = 18,325
%
Cases n1b
Cases n1b
(95% CI)f
Surveillance timec (n2d)
Surveillance timec (n2d)
d. n2 = Number of participants at risk for the endpoint.
e. No confirmed cases were identified in adolescents 12 to 15 years of age.
f. Two-sided confidence interval (CI) for vaccine efficacy (VE) is derived based on the Clopper and
Pearson method adjusted to the surveillance time. CI not adjusted for multiplicity.
In the second primary analysis, efficacy of Comirnaty (tozinameran) in preventing first
COVID-19 occurrence from 7 days after Dose 2 compared to placebo was 94.6% (95% credible
interval of 89.9% to 97.3%) in participants 16 years of age and older with or without evidence
of prior infection with SARS-CoV-2.
Additionally, subgroup analyses of the primary efficacy endpoint showed similar efficacy point
estimates across genders, ethnic groups, and participants with medical comorbidities associated
with high risk of severe COVID-19.
Updated efficacy analyses were performed with additional confirmed COVID-19 cases accrued
during blinded placebo-controlled follow-up through 13 March 2021, representing up to
6 months of follow-up after Dose 2 for participants in the efficacy population.
The updated vaccine efficacy information is presented in Table 16.
Table 16: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2, by age
subgroup – participants without evidence of infection prior to 7 days after Dose 2 –
evaluable efficacy (7 days) population during the placebo-controlled follow-up period
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
of prior SARS-CoV-2 infection*
Comirnaty
(tozinameran)
Placebo
Na=20,998
Na=21,096
Cases n1b
Cases n1b
Vaccine efficacy %
Subgroup
Surveil ance Timec (n2d) Surveil ance Timec (n2d)
(95% CIe)
All participantsf
77
850
91.3
6.247 (20,712)
6.003 (20,713)
(89.0, 93.2)
16 to 64 years
70
710
90.6
4.859 (15,519)
4.654 (15,515)
(87.9, 92.7)
65 years and older
7
124
94.5
1.233 (4192)
1.202 (4226)
(88.3, 97.8)
65 to 74 years
6
98
94.1
0.994 (3350)
0.966 (3379)
(86.6, 97.9)
75 years and older
1
26
96.2
0.239 (842)
0.237 (847)
(76.9, 99.9)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative
at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 25 of 41
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Two-sided confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method
adjusted to the surveillance time.
f. Included confirmed cases in participants 12 to 15 years of age: 0 in the Comirnaty (tozinameran) group (both
without and with or without evidence of prior SARS-CoV-2 infection); 16 and 18 in the placebo group
(without and with or without evidence of prior SARS-CoV-2 infection, respectively).
Efficacy against severe COVID-19 in participants 12 years of age or older – after 2 doses
As of 13 March 2021, vaccine efficacy against severe COVID-19 is presented only for
participants with or without prior SARS-CoV-2 infection (Table 17) as the COVID-19 case
counts in participants without prior SARS-CoV-2 infection were the same as those in
participants with or without prior SARS-CoV-2 infection in both the Comirnaty (tozinameran)
and placebo groups.
Table 17. Vaccine Efficacy – First Severe COVID-19 Occurrence in Participants With
or Without* Prior SARS-CoV-2 Infection Based on Food and Drug Administration
(FDA)† Definition After Dose 1 or From 7 Days After Dose 2 in the Placebo-Controlled
Follow-up
Comirnaty (tozinameran)
Placebo
Cases n1a
Cases n1a
Vaccine Efficacy %
Surveil ance Time (n2b) Surveil ance Time (n2b)
(95% CIc)
1
30
96.7
After Dose 1d
8.439e (22,505)
8.288e (22,435)
(80.3, 99.9)
1
21
95.3
7 days after Dose 2f
6.522g (21,649)
6.404g (21,730)
(70.9, 99.9)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at
least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or increased
shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat; diarrhoea;
vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative
at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
† Severe illness from COVID-19 as defined by FDA is confirmed COVID-19 and presence of at least 1 of the
following:
• Clinical signs at rest indicative of severe systemic illness (respiratory rate ≥30 breaths per minute, heart
rate ≥125 beats per minute, saturation of oxygen ≤93% on room air at sea level, or ratio of arterial
oxygen partial pressure to fractional inspired oxygen <300 mm Hg);
• Respiratory failure [defined as needing high-flow oxygen, noninvasive ventilation, mechanical
ventilation or extracorporeal membrane oxygenation (ECMO)];
• Evidence of shock (systolic blood pressure <90 mm Hg, diastolic blood pressure <60 mm Hg, or
requiring vasopressors);
• Significant acute renal, hepatic, or neurologic dysfunction;
• Admission to an Intensive Care Unit;
• Death.
a. n1 = Number of participants meeting the endpoint definition.
b. n2 = Number of participants at risk for the endpoint.
c. Two-side confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method
adjusted to the surveillance time.
d. Efficacy assessed based on the Dose 1 all available efficacy (modified intention-to-treat) population that
included all randomised participants who received at least 1 dose of study intervention.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 26 of 41
e. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from Dose 1 to the end of the surveillance
period.
f. Efficacy assessed based on the evaluable efficacy (7 Days) population that included al eligible randomised
participants who receive all dose(s) of study intervention as randomised within the predefined window, have
no other important protocol deviations as determined by the clinician
g. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
Efficacy and immunogenicity in adolescents 12 to 15 years of age – after 2 doses
An analysis of Study C4591001 has been performed in adolescents 12 to 15 years of age up to
a data cutoff date of 13 March 2021.
The vaccine efficacy information in adolescents 12 to 15 years of age is presented in Table 18.
Table 18: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2 –
participants without evidence of infection and with or without evidence of infection prior
to 7 days after Dose 2 – adolescents 12 to 15 years of age evaluable efficacy (7 days)
population
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
without evidence of prior SARS-CoV-2 infection*
Comirnaty
(tozinameran)
Placebo
Na = 1005
Na = 978
Cases n1b
Cases n1b
Vaccine efficacy
Surveil ance timec (n2d) Surveil ance timec (n2d)
% (95% CIe)
Adolescents
0
16
12 to 15 years
0.154 (1001)
0.147 (972)
100.0 (75.3, 100.0)
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
with or without* evidence of prior SARS-CoV-2 infection
Comirnaty
(tozinameran)
Placebo
Na = 1119
Na = 1110
Cases n1b
Cases n1b
Vaccine efficacy
Surveil ance timec (n2d) Surveil ance timec (n2d)
% (95% CIe)
Adolescents
0
18
12 to 15 years
0.170 (1109)
0.163 (1094)
100.0 (78.1, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at
least 1 symptom consistent with COVID-19 [*Case definition: (at least 1 of) fever, new or increased cough, new
or increased shortness of breath, chills, new or increased muscle pain, new loss of taste or smell, sore throat,
diarrhoea or vomiting).
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the last dose) of
past SARS-CoV-2 infection (i.e, N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not
detected by nucleic acid amplification tests (NAAT) [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all subjects within each group at
risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of subjects at risk for the endpoint.
e. Confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method adjusted
for surveillance time. CI not adjusted for multiplicity.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 27 of 41
In Study C4591001 an analysis of SARS-CoV-2 neutralising titres in a randomly selected
subset of participants was performed to demonstrate non-inferior immune responses (within
1.5-fold) comparing adolescents 12 to 15 years of age to participants 16 to 25 years of age who
had no serological or virological evidence of past SARS-CoV-2 infection. The immune
response to Comirnaty (tozinameran) in adolescents 12 to 15 years of age (n = 190) was non-
inferior to the immune response in participants 16 to 25 years of age (n = 170), based on results
for SARS-CoV-2 neutralising titres at 1 month after Dose 2. The geometric mean titres (GMT)
ratio of the adolescents 12 to 15 years of age group to the participants 16 to 25 years of age
group was 1.76, with a 2-sided 95% CI of 1.47 to 2.10, meeting the 1.5-fold non-inferiority
criterion (the lower bound of the 2-sided 95% CI for the geometric mean ratio [GMR] >0.67),
which indicates a statistically greater response in the adolescents 12 to 15 years of age than that
of participants 16 to 25 years of age.
An updated efficacy analysis of Study C4591001 has been performed in approximately 2,260
adolescents 12 to 15 years of age evaluating confirmed COVID-19 cases accrued up to a data
cut-off date of 2 September 2021, representing up to 6 months of follow-up after Dose 2 for
participants in the efficacy population.
The updated vaccine efficacy information in adolescents 12 to 15 years of age is presented in
Table 19.
Table 19: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Dose 2:
Without Evidence of Infection and With or Without Evidence of Infection Prior to 7 Days
After Dose 2 – Blinded Placebo-Controlled Follow-up Period, Adolescents 12 To 15 Years
of Age Evaluable Efficacy (7 Days) Population
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
without evidence of prior SARS-CoV-2 infection*
Comirnaty
(tozinameran)
Placebo
Na=1057
Na=1030
Vaccine Efficacy
Cases n1b
Cases n1b
%
Surveil ance Timec (n2d) Surveil ance Timec (n2d)
(95% CIe)
Adolescents
0
28
100.0
12 to 15 years of age
0.343 (1043)
0.322 (1019)
(86.8, 100.0)
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of
age with or without evidence of prior SARS-CoV-2 infection
Comirnaty
(tozinameran)
Placebo
Na=1119
Na=1109
Vaccine Efficacy
Cases n1b
Cases n1b
%
Surveil ance Timec (n2d) Surveil ance Timec (n2d)
(95% CIe)
Adolescents
0
30
100.0
12 to 15 years of age
0.362 (1098)
0.345 (1088)
(87.5, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum]
negative at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had
negative NAAT (nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the
analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 28 of 41
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each
group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the
end of the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Two-sided confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson
method adjusted for surveillance time.
Efficacy in children 5 to 11 years of age – after 2 doses
An initial descriptive efficacy analysis of Study C4591007 has been performed in 1,968
children 5 to 11 years of age without evidence of infection prior to 7 days after Dose 2. This
analysis evaluated confirmed symptomatic COVID-19 cases accrued up to a data cut-off date
of 8 October 2021.
The initial descriptive vaccine efficacy results in children 5 to 11 years of age without evidence
of prior SARS-CoV-2 infection are presented in Table 20. None of the cases accrued met
criteria for severe COVID-19 or multisystem inflammatory syndrome in children (MIS-C). No
cases of COVID-19 were observed in either the vaccine group or the placebo group in
participants with evidence of prior SARS-CoV-2 infection.
Table 20: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Dose 2:
Without Evidence of Infection Prior to 7 Days After Dose 2 – Phase 2/3 – Children 5 To
11 Years of Age Evaluable Efficacy Population
First COVID-19 occurrence from 7 days after Dose 2 in children 5 to 11 years of age
without evidence of prior SARS-CoV-2 infection*
Comirnaty±
(tozinameran)
10 micrograms/dose
Placebo
Na=1305
Na=663
Vaccine Efficacy
Cases n1b
Cases n1b
%
Surveil ance Timec (n2d)
Surveil ance Timec (n2d)
(95% CI)
Children 5 to
3
16
90.7
11 years of age
0.322 (1273)
0.159 (637)
(67.7, 98.3)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum]
negative at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had
negative NAAT (nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the
analysis.
± Pfizer-BioNTech COVID-19 Vaccine (10 micrograms modRNA).
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each
group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the
end of the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
Prespecified hypothesis-driven efficacy analysis was performed with additional confirmed
COVID-19 cases accrued during blinded placebo-controlled follow-up, representing up to 6
months after Dose 2 in the efficacy population.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 29 of 41
In the efficacy analysis of Study C4591007 in children 5 to 11 years of age without evidence
of prior infection, there were 10 cases out of 2,703 participants who received the vaccine and
42 cases out of 1,348 participants who received placebo. The point estimate for efficacy is
88.2% (95% CI: 76.2, 94.7). In participants with or without evidence of prior infection there
were 12 cases in the 3,018 who received vaccine and 42 cases in 1,511 participants who
received placebo. The point estimate for efficacy is 85.7% (95% CI: 72.4, 93.2).
Immunogenicity in children 5 to 11 years of age – after 2 doses
Study C4591007 is a Phase 1/2/3 study comprised of an open-label vaccine dose-finding
portion (Phase 1) and a multicentre, multinational, randomised, saline placebo-controlled,
observer-blind efficacy portion (Phase 2/3) that has enrolled participants 5 to 11 years of age.
In C4591007, an analysis of SARS-CoV-2 50% neutralising titres (NT50) 1 month after Dose
2 in a randomly selected subset of participants demonstrated effectiveness by immunobridging
of immune responses comparing children 5 to 11 years of age in the Phase 2/3 part of Study
C4591007 to participants 16 to 25 years of age in the Phase 2/3 part of Study C4591001 who
had no serological or virological evidence of past SARS-CoV-2 infection up to 1 month after
Dose 2, meeting the prespecified immunobridging criteria for both the geometric mean ratio
(GMR) and the seroresponse difference with seroresponse defined as achieving at least 4-fold
rise in SARS-CoV-2 NT50 from baseline (before Dose 1).
The ratio of the SARS-CoV-2 NT50 in children 5 to 11 years of age to that of young adults 16
to 25 years of age was 1.04 (2-sided 95% CI: 0.93, 1.18), as presented in Table 21.
Table 21: Summary of geometric mean ratio for 50% neutralising titre – Comparison
of children 5 to 11 years of age (Study C4591007) to participants 16 to 25 years of age
(Study C4591001) – participants without* evidence of infection up to 1 month after Dose
2 – evaluable immunogenicity population
Comirnaty (tozinameran)
5 to 11 years/
10 microgram/dose 30 microgram/dose
16 to 25 years
5 to 11 years
16 to 25 years
na=264
na=253
Met
Assay
Time
GMTc
GMTc
GMRd immunobridging
pointb
(95% CIc)
(95% CIc)
(95% CId)
objectivee
(Y/N)
SARS-CoV-2
neutralisation 1 month
1197.6
1146.5
1.04
assay - NT50 after
(1106.1, 1296.6)
(1045.5, 1257.2) (0.93, 1.18)
Y
(titre)f
Dose 2
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre;
LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test; NT50 = 50% neutralising titre;
SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
*Participants who had no serological or virological evidence (up to 1 month post-Dose 2 blood sample collection)
of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and 1 month after Dose
2, SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2, and negative NAAT (nasal swab) at
any unscheduled visit up to 1 month after Dose 2 blood collection) and had no medical history of COVID-
19 were included in the analysis.
a. n = Number of participants with valid and determinate assay results for the specified assay at the given
dose/sampling time point.
b. Protocol-specified timing for blood sample collection.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 30 of 41
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
d. GMRs and 2-sided 95% CIs were calculated by exponentiating the mean difference of the logarithms of the
titres (Group 1[5 to 11 years of age] - Group 2 [16 to 25 years of age]) and the corresponding CI (based on
the Student t distribution).
e. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67 and
the point estimate of the GMR is ≥0.8.
f. SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation
Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus
neutralisation is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution
at which 50% of the virus is neutralised.
Among participants without prior evidence of SARS-CoV-2 infection up to 1 month after
Dose 2, 99.2% of children 5 to 11 years of age and 99.2% of participants 16 to 25 years of age
had a seroresponse from before vaccination to 1 month after Dose 2. The difference in
proportions of participants who had seroresponse between the 2 age groups (children – young
adult) was 0.0% (2-sided 95% CI: -2.0%, 2.2%) as presented in Table 22.
Table 22: Difference in percentages of participants with seroresponse – participants
without evidence of infection up to 1 month after Dose 2 – immunobridging subset –
Phase 2/3 – comparison of 5 to 11 years of age to Study C4591001 Phase 2/3 16 to 25 years
of age – evaluable immunogenicity population
Comirnaty (tozinameran)
10
30
5 to 11 years/
microgram/dose microgram/dose
16 to 25 years
5 to 11 years
16 to 25 years
Na=264
Na=253
Met
Assay
Time
nc (%)
nc (%)
Difference %e immunobridging
pointb
(95% CId)
(95% CId)
(95% CIf)
objectiveg
(Y/N)
SARS-CoV-2
neutralisation
1 month
262 (99.2)
251 (99.2)
0.0
assay – NT50 after
(97.3, 99.9)
(97.2, 99.9)
(-2.0, 2.2)
Y
(titre)h
Dose 2
Abbreviations: LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test;
N-binding = SARS-CoV-2 nucleoprotein–binding; NT50 = 50% neutralising titre 50; SARS-CoV-2 = severe acute
respiratory syndrome coronavirus 2.
Note: Seroresponse is defined as achieving a ≥4-fold rise from baseline (before Dose 1). If the baseline measurement
is below the LLOQ, a postvaccination assay result ≥4 × LLOQ is considered a seroresponse.
Note: Participants who had no serological or virological evidence (up to 1 month post-Dose 2 blood sample
collection) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and 1 month after
Dose 2, SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2, and negative NAAT (nasal swab) at
any unscheduled visit up to 1 month after Dose 2 blood collection) and had no medical history of COVID-19 were
included in the analysis.
a. N = number of participants with valid and determinate assay results both before vaccination and at 1 month after
Dose 2. These values are the denominators for the percentage calculations.
b. Protocol-specified timing for blood sample collection.
c. n = Number of participants with seroresponse for the given assay at the given dose/sampling time point.
d. Exact 2-sided CI based on the Clopper and Pearson method.
e. Difference in proportions, expressed as a percentage (Group 1 [5 to 11 years of age] – Group 2 [16 to 25 years
of age]).
f. 2-Sided CI, based on the Miettinen and Nurminen method for the difference in proportions, expressed as a
percentage.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 31 of 41
g. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the difference in proportions is greater
than -10.0%.
h. SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation Assay.
The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus neutralisation is
read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution at which 50% of
the virus is neutralised.
Immunogenicity in participants 18 years of age and older – after booster dose
Effectiveness of a booster dose of Comirnaty (tozinameran) was based on an assessment of
50% neutralising titres (NT50) against SARS-CoV-2 (USA_WA1/2020). In Study C4591001,
analyses of NT50 1 month after the booster dose compared to 1 month after the primary series
in individuals 18 to 55 years of age who had no serological or virological evidence of past
SARS-CoV-2 infection up to 1 month after the booster vaccination demonstrated
noninferiority for both GMR and difference in seroresponse rates. Seroresponse for a
participant was defined as achieving a ≥4-fold rise in NT50 from baseline (before Dose 1),
These analyses are summarised in Table 23.
Table 23. SARS-CoV-2 neutralisation assay - NT50 (titre)† (SARS-CoV-2
USA_WA1/2020) – GMT and seroresponse rate comparison of 1 month after booster dose
to 1 month after primary series – participants 18 to 55 years of age without evidence of
infection up to 1 month after booster dose* – booster dose evaluable immunogenicity
population±
1 month after
booster dose/-
1 month after
Met
1 month after
1 month after
primary
noninferiority
booster dose
primary series
series
objective
n
(95% CI)
(95% CI)
(97.5% CI)
(Y/N)
Geometric mean
50% neutralising
2466.0b
755.7
b
3.26c
titre (GMTb)
212a (2202.6, 2760.8) (663.1, 861.2)
(2.76, 3.86)
Yd
Seroresponse rate
199f
190f
(%) for 50%
99.5%
95.0%
4.5%g
neutralising titre† 200e (97.2%, 100.0%) (91.0%, 97.6%) (1.0%, 7.9%
h)
Yi
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre;
LLOQ = lower limit of quantitation; N-binding = SARS-CoV-2 nucleoprotein-binding; NAAT = nucleic acid
amplification test; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute respiratory syndrome
coronavirus 2; Y/N = yes/no.
† SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation
Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus
neutralisation is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum
dilution at which 50% of the virus is neutralised.
* Participants who had no serological or virological evidence (up to 1 month after receipt of a booster dose
of Comirnaty(tozinameran)) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative
and SARS-CoV-2 not detected by NAAT [nasal swab]) and had a negative NAAT (nasal swab) at any
unscheduled visit up to 1 month after the booster dose were included in the analysis.
± All eligible participants who had received 2 doses of Comirnaty (tozinameran) as initially randomised,
with Dose 2 received within the predefined window (within 19 to 42 days after Dose 1), received a booster
dose of Comirnaty (tozinameran), had at least 1 valid and determinate immunogenicity result after booster
dose from a blood collection within an appropriate window (within 28 to 42 days after the booster dose),
and had no other important protocol deviations as determined by the clinician.
a. n = Number of participants with valid and determinate assay results at both sampling time points within
specified window.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 32 of 41
b. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
c. GMRs and 2-sided 97.5% CIs were calculated by exponentiating the mean differences in the logarithms of
the assay and the corresponding CIs (based on the Student t distribution).
d. Noninferiority is declared if the lower bound of the 2-sided 97.5% CI for the GMR is > 0.67 and the point
estimate of the GMR is ≥ 0.80.
e. n = Number of participants with valid and determinate assay results for the specified assay at baseline,
1 month after Dose 2 and 1 month after the booster dose within specified window. These values are the
denominators for the percentage calculations.
f. Number of participants with seroresponse for the given assay at the given dose/sampling time point. Exact
2-sided CI based on the Clopper and Pearson method.
g. Difference in proportions, expressed as a percentage (1 month after booster dose – 1 month after Dose 2).
h. Adjusted Wald 2-sided CI for the difference in proportions, expressed as a percentage.
i. Noninferiority is declared if the lower bound of the 2-sided 97.5% CI for the percentage difference is
> -10%.
Relative vaccine efficacy in participants 16 years of age and older – after booster dose
An interim efficacy analysis of Study C4591031, a placebo-controlled booster study, was
performed in approximately 10,000 participants 16 years of age and older who were recruited
from Study C4591001, evaluated confirmed COVID-19 cases accrued from at least 7 days after
booster vaccination up to a data cut-off date of 8 February 2022 (a period when Delta and then
Omicron was the predominant variant), which represents a median of 2.8 months (range 0.3 to
7.5 months) post-booster follow-up. Vaccine efficacy of the Comirnaty (tozinameran) booster
dose after the primary series relative to the placebo booster group who only received the
primary series dose was assessed. The relative vaccine efficacy information for participants
16 years of age and older is presented in Table 24.
Table 24: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Booster
Vaccination – Participants 16 Years of Age and Older Without Evidence of Infection and
Participants With or Without Evidence of Infection Prior to 7 Days After Booster
Vaccination – Evaluable Efficacy Population
First COVID-19 occurrence from 7 days after booster dose in participants without evidence
of prior SARS-CoV-2 infection*
Comirnaty (tozinameran)
Placebo
Relative
Na=4689
Na=4664
Vaccine
Cases n1b
Cases n1b
Efficacye %
Surveil ance Timec (n2d)
Surveil ance Timec (n2d)
(95% CIf)
First COVID-19
occurrence from
7 days after booster
63
148
63.9
vaccination
1.098 (4639)
0.932 (4601)
(51.1, 73.5)
First COVID-19 occurrence from 7 days after booster dose in participants with or without
evidence of prior SARS-CoV-2 infection
Comirnaty (tozinameran)
Placebo
Relative
Na=4977
Na=4942
Vaccine
Cases n1b
Cases n1b
Efficacye %
Surveil ance Timec (n2d)
Surveil ance Timec (n2d)
(95% CIf)
First COVID-19
occurrence from
7 days after booster
67
150
62.4
vaccination
1.173 (4903)
0.989 (4846)
(49.5, 72.2)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 33 of 41
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the booster
vaccination) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and
SARS-CoV-2 not detected by NAAT [nasal swab] at Visit 1, and had a negative NAAT [nasal swab] at
any unscheduled visit prior to 7 days after booster vaccination) were included in the analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each
group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after the booster
vaccination to the end of the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Relative vaccine efficacy of the Comirnaty (tozinameran) booster group relative to the placebo group
(non-booster).
f. Two-sided confidence interval (CI) for relative vaccine efficacy is derived based on the Clopper and
Pearson method adjusted for surveillance time.
Immunogenicity in children 5 to 11 years of age – after booster dose
Effectiveness of a booster dose of Comirnaty (tozinameran) was based on an assessment of
NT50 against the reference strain of SARS-CoV-2 (USA_WA1/2020). Analyses of NT50
1 month after the booster dose compared to before the booster dose demonstrated a substantial
increase in GMTs in individuals 5 to 11 years of age who had no serological or virological
evidence of past SARS-CoV-2 infection up to 1 month after the booster dose. This analysis is
summarised in Table 25.
Table 25: Summary of Geometric Mean Titres – NT50 – Participants Without
Evidence of Infection – Phase 2/3 – Immunogenicity Set – 5 to 11 Years of Age – Evaluable
Immunogenicity Population
Comirnaty (tozinameran) 10 mcg/Dose
3-Dose Set
2-Dose Set
Total
Dose/
Sampling
GMTc
GMTc
GMTc
Assay
Time Pointa
nb
(95% CIc)
nb
(95% CIc) nb
(95% CIc)
20.5
20.5
20.5
1 month Prevax 79
(20.5, 20.5)
67 (20.5, 20.5) 146 (20.5, 20.5)
SARS-CoV-2 1 month after Dose
1659.4
1110.7
1253.9
neutralisation
2
29 (1385.1, 1988.0) 67 (965.3, 1278.1) 96 (1116.0, 1408.9)
assay - NT50
271.0
271.0
(titre)
3 months Prevax 67 (229.1, 320.6) -
-
67 (229.1, 320.6)
1 month after Dose
2720.9
2720.9
3
67 (2280.1, 3247.0) -
-
67 (2280.1, 3247.0)
Abbreviations: CI = confidence interval; GMT = geometric mean titre; LLOQ = lower limit of quantitation;
NAAT = nucleic acid amplification test; N-binding = SARS-CoV-2 nucleoprotein–binding; NT50 = 50%
neutralising titre; Prevax = before vaccination; SARS-CoV-2 = severe acute respiratory syndrome coronavirus
2.
Note: Three-dose immunogenicity set included the first 130 participants who received Dose 3 and completed
1-month post–Dose 3 visit prior to March 15, 2022. Among those, 30 had blood sample collection at 1-month
post-Dose 2. Two-dose immunogenicity set included an extra 67 participants randomly selected from previous
Dose-2 evaluable immunogenicity population and without evidence of infection up to 1-month post–Dose 2
subset used for 2-dose immunobridging analysis.
Note: Participants included in this analysis had no serological or virological evidence of past SARS-CoV-2
infection up to the 1-month post–Dose 2 (for 1-month post–Dose 2 time point) or 1-month post–Dose 3 (for pre–
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 34 of 41
Dose 3 and 1-month post–Dose 3 time point) study blood sample collection. Having no evidence of past SARS-
CoV-2 infection up to 1-month post–Dose 2 was defined as having a negative N-binding antibody (serum) result
at the Dose 1 and 1-month post–Dose 2 study visits; a negative NAAT (nasal swab) result at the Dose 1 and
Dose 2 study visits and any unscheduled visit prior to the 1-month post–Dose 2 blood sample collection; and no
medical history of COVID-19. Having no evidence of past SARS-CoV-2 infection up to 1-month post-Dose 3
was defined as having a negative N-binding antibody (serum) result at the Dose 1, 1-month post–Dose 2 (if
available), Dose 3, and 1-month post–Dose 3 study visits; a negative NAAT (nasal swab) result at the Dose 1,
Dose 2, and Dose 3 study visits and any unscheduled visit prior to the 1-month post–Dose 3 blood sample
collection; and no medical history of COVID-19.
a. Protocol-specified timing for blood sample collection.
b. n = Number of participants with valid and determinate assay results for the specified assay at the given
dose/sampling time point.
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 ×
LLOQ.
Immunogenicity in children 5 to 11 years of age on the Omicron variant (B1.1.529) –
after booster dose
The neutralising GMTs against both the Omicron variant (B1.1.529) and reference strain were
substantially increased after booster vaccination compared with after the 2-dose primary series.
At 1-month post-Dose 2, the observed neutralising GMTs for the Omicron variant (B1.1.529)
and reference strain were 27.6 and 323.8, respectively. At 1-month post-Dose 3, the observed
neutralising GMTs for the Omicron variant (B1.1.529) and reference strain were 614.4 and
1702.8, respectively (see Table 26).
For the Omicron variant (B1.1.529), neutralising titres after booster vaccination (1-month post-
Dose 3) increased 22-fold over those after the 2-dose primary series (1-month post-Dose 2).
For the reference strain, the increase after the booster relative to the primary series was 5.3-
fold.
Table 26: Summary of Geometric Mean Titres – Omicron-Neutralisation Subset –
Participants Without Evidence of Infection – Phase 2/3 – Immunogenicity Set – 5 to 11
Years of Age – Evaluable Immunogenicity Population
Comirnaty (tozinameran)
10 mcg/Dose
Vaccine Group (as Randomised)
GMTc
Assay
Time Pointb
nb
(95% CIc)
SARS-COV-2 FFRNT-
27.6
B.1.1.529 strain
1 month after Dose 2
29
(22.1, 34.5)
(Omicron) - NT50
614.4
(titre)
1 month after Dose 3
17
(410.7, 919.2)
SARS-CoV-2 FFRNT-
323.8
reference strain - NT50
1 month after Dose 2
29
(267.5, 392.1)
(titre)
1702.8
1 month after Dose 3
17
(1282.6, 2260.7)
Abbreviations: CI = confidence interval; FFRNT = fluorescence focus reduction neutralisation test;
GMT = geometric mean titre; LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test;
N-binding = SARS-CoV-2 nucleoprotein–binding; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute
respiratory syndrome coronavirus 2.
Note: Participants included in this analysis had no serological or virological evidence of past SARS-CoV-2
infection up to the 1-month post–Dose 2 (for 1-month post–Dose 2 time point) or 1-month post–Dose 3 (for
1-month post–Dose 3 time point) study blood sample collection. Having no evidence of past SARS-CoV-2
infection up to 1-month post–Dose 2 was defined as having a negative N-binding antibody (serum) result at
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 35 of 41
the Dose 1 and 1-month post–Dose 2 study visits; a negative NAAT (nasal swab) result at the Dose 1 and Dose
2 study visits and any unscheduled visit prior to the 1-month post–Dose 2 blood sample collection; and no
medical history of COVID-19. Having no evidence of past SARS-CoV-2 infection up to 1-month post–Dose 3
was defined as having a negative N-binding antibody (serum) result at the Dose 1, 1-month post–Dose 2 (if
available), Dose 3, and 1-month post–Dose 3 study visits; a negative NAAT (nasal swab) result at the Dose 1,
Dose 2, and Dose 3 study visits and any unscheduled visit prior to the 1-month post–Dose 3 blood sample
collection; and no medical history of COVID-19.
a. Protocol-specified timing for blood sample collection.
b. n = Number of participants with valid and determinate assay results for the specified assays at the given
dose/sampling time point.
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
Immunogenicity in immunocompromised participants (adults and children)
Study C4591024 is a Phase 2b, open-label study (n=124) that enrolled immunocompromised
participants 2 to 17 years of age receiving immunomodulator therapy or who have undergone
solid organ transplant (within the previous 3 months) and are on immunosuppression or who
have undergone bone marrow or stem cell transplant at least 6 months prior to enrollment.
Study C4591024 also enrolled immunocompromised participants 18 years of age and older
treated for NSCLC or CLL, receiving hemodialysis for secondary to end-stage renal disease,
or receiving immunomodulator therapy for an autoimmune inflammatory disorder. Study
participants did not have a past clinical or microbiological diagnosis of COVID-19. Participants
received 4 age-appropriate doses of Comirnaty (tozinameran) (3 micrograms, 10 micrograms,
or 30 micrograms); the first 2 doses separated by 21 days, with the third dose occurring 28 days
after the second dose, followed by a fourth dose, 3 to 6 months after Dose 3.
The immunogenicity results pre-vaccination and after 3 and 4 doses of Comirnaty
(tozinameran) in immunocompromised participants 2 years of age and older are presented in
Table 27.
Table 27. Summary of Geometric Mean Titres – Participants With or Without
Evidence of Infection by Age Group – All-Available Immunogenicity
Population
Comirnaty (tozinameran)
3 micrograms
10 micrograms
30 micrograms 30 micrograms
Age Group:
Age Group:
Age Group:
Age Group:
2 to 4 Years
5 to 11 Years
12 to 17 Years
≥
18 Years
Dose/
Sampling
GMTc
GMTc
GMTc
GMTc
Assay
Time Pointb nc
(95% CId)
nc (95% CId) nc
(95% CId) nc (95% CId)
SARS-CoV-2
44.8
44.5
54.2
82.2
neutralisation
1/Prevax
32
(42.2, 47.7)
62 (42.5, 46.5) 14 (33.7, 87.0) 6 (16.0, 422.5)
assay –
1566.5
2940.6
787.1
reference strain
942.3
(1019.9,
(1175.5,
(66.5,
– NT50 (titre)a 3/1 Month 32 (537.1, 1653.4) 60
2405.9)
14
7356.0)
6
9321.5)
922.2
3284.5
606.2
487.8
(586.7,
(1609.8,
(5.3,
4/Pre-Dose 4 29
(269.0, 884.9) 57
1449.3)
11
6701.3)
3
68756.0)
6463.4
13457.1
1031.3
3447.0
(4319.7,
(5270.1,
(56.9,
4/1 Month 26 (1851.0, 6419.2) 50
9670.9)
9
34362.4)
4
18681.7)
2382.3
5776.1
1605.6
1296.7
(1554.3,
(2801.4,
(28.5,
4/6 Months 25 (674.2, 2494.0) 49
3651.2)
8
11909.2)
3
90614.9)
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 36 of 41
Abbreviations: CI = confidence interval; GMT = geometric mean titre; LLOQ = lower limit of quantitation; NT50 = 50%
neutralising titre; Prevax = before vaccination; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
a. SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (original strain [USA-WA1/2020, isolated
in January 2020]).
b Protocol-specified timing for blood sample collection.
c. n = Number of participants with valid and determinate assay results for the specified assay at the given dose/sampling time
point.
d. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the corresponding CIs
(based on the Student t distribution). Assay results below the LLOQ were set to 0.5 × LLOQ.
Analysis of immunogenicity data at 1 month after Dose 3 (32 participants 2 to 4 years of age,
60 participants 5 to 11 years of age, 14 participants 12 to 17 years of age, and 6 participants
18 years of age and older) and 1 month after Dose 4 (26 participants 2 to 4 years of age, 50
participants 5 to 11 years of age, 9 participants 12 to 17 years of age, and 4 participants 18
years of age and older) in the all available immunogenicity population with or without evidence
of prior infection demonstrated a vaccine-elicited immune response.
GMTs were observed to be substantially higher at 1 month after Dose 3 and further increased
at 1 month after Dose 4 and remained high at 6 months after Dose 4 compared to levels
observed before study vaccination across age groups and disease subsets.
5.2 Pharmacokinetic properties
Not applicable.
5.3 Preclinical safety data
Genotoxicity/Carcinogenicity
Neither genotoxicity nor carcinogenicity studies were performed. The components of
Comirnaty (lipids and mRNA) are not expected to have genotoxic potential.
6. PHARMACEUTICAL PARTICULARS
6.1 List of excipients
((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC)
Cholesterol
Trometamol
Trometamol hydrochloride
Sucrose
Water for injections
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 37 of 41
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those
mentioned in Section 6.6 Special precautions for disposal and other handling.
6.3 Shelf life
Unopened vial
Frozen vial
18 months when stored at -90°C to -60°C.
The vaccine will be received frozen at -90°C to -60°C. Frozen vaccine can be stored either at
-90°C to -60°C or 2°C to 8°C upon receipt.
For thawing instructions of the frozen vials, see Section 6.6 Special precautions for disposal
and other handling.
Thawed vial
If the vaccine is received at 2°C to 8°C it should be stored at 2°C to 8°C. Once removed from
frozen storage, the unopened vial may be stored refrigerated at 2°C to 8°C for a single period
of up to 10 weeks within the 18 month shelf life.
Upon moving the product to 2°C to 8°C storage, the updated expiry date must be written on
the outer carton and the vaccine should be used or discarded by the updated expiry date. The
original expiry date should be crossed out.
Check that the expiry date on the outer carton and/or vial has been updated to reflect the
refrigerated expiry date and that the original expiry date has been crossed out.
Prior to use, the unopened vials can be stored for up to 12 hours at temperatures between
8ºC to 30ºC.
Thawed vials can be handled in room light conditions.
Once thawed the vaccine should not be re-frozen.
Diluted medicinal product
Chemical and physical in-use stability has been demonstrated for 12 hours at 2ºC to 30°C, after
dilution with sodium chloride 9 mg/mL (0.9%) solution for injection. From a microbiological
point of view, unless the method of dilution precludes the risk of microbial contamination, the
product should be used immediately. If not used immediately, in-use storage times and
conditions are the responsibility of the user.
6.4 Special precautions for storage
Check that the expiry date has been updated to reflect the refrigerated EXP date and that the
original expiry date has been crossed out.
Store in the original package to protect from light. During storage, minimise exposure to room
light, and avoid exposure to direct sunlight and ultraviolet light.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 38 of 41
For detailed instructions see Section 6.6 Special precautions for disposal and other handling.
Once thawed, the vaccine cannot be re-frozen.
Thawed vials can be handled in room light conditions.
For storage conditions after thawing and dilution of the medicinal product, see Section 6.3
Shelf life.
For additional advice on storing Comirnaty LP.8.1, contact Pfizer New Zealand on 0800 736
363.
6.5 Nature and contents of container
Comirnaty LP.8.1 (Yellow cap, must dilute) 0.48 mL fill volume in 2 mL clear multidose vial
(Type I glass) with a stopper (synthetic bromobutyl rubber) and a yellow flip-off plastic cap
with aluminium seal. Each vial contains 3 doses of 0.3 mL after dilution, see Section 6.6
Special precautions for disposal and other handling.
Pack size: 10 vials
6.6 Special precautions for disposal and other handling
Comirnaty LP.8.1 Concentrate for suspension for injection (Yellow cap)
Handling Instructions
Handing prior to use
Frozen vials must be completely thawed prior to use. Frozen vials should be transferred to 2 °C
to 8 °C to thaw. Thaw times for 10-vial packs are noted in table below:
Vial Cap Color
Time That May Be Required For a 10-vial Pack
to Thaw (at 2 °C to 8 °C)
Yellow
2 hours
• Upon moving frozen vaccine to 2 °C to 8 °C storage, update the expiry date on the carton.
The updated expiry date should reflect 10 weeks from the date of transfer to refrigerated
conditions (2 °C to 8 °C) and not exceeding the original printed expiry date (EXP).
• Alternatively, individual frozen vials may be thawed for 30 minutes at temperatures up to
30 °C for immediate use.
• If the vaccine is received at 2 °C to 8 °C it should continue to be stored at 2 °C to 8 °C.
Check that the carton has been previously updated to reflect the 10-week refrigerated expiry
date.
• Unopened vials can be stored for up to 12 hours at temperatures up to 30 °C. Total storage
time between 8 ºC to 30 ºC, inclusive of storage before and after puncture, should not
exceed 24 hours.
Comirnaty LP.8.1 – Concentrate for Suspension for Injection (Yellow cap)
Preparation for administration
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 39 of 41
Comirnaty LP.8.1 Concentrate for suspension for injection should be prepared by a healthcare
professional using aseptic technique to ensure the sterility of the prepared diluted suspension.
Vials of Comirnaty LP.8.1 Concentrate for suspension for injection have a Yellow cap and
requires dilution.
Vial verification
Prior to administration, check the name and strength of the vaccine on the vial label and the
colour of the vial cap and vial label border to ensure it is the intended presentation. Check
whether the vial is a single dose vial or a multidose vial and check if the vial requires dilution.
Prior to dilution
• After the thawed vial has reached room temperature, gently invert it 10 times prior to
dilution.
Do not shake.
• Check appearance of vaccine.
o
Yellow cap vials: Prior to dilution, the vaccine is a clear to slightly opalescent
solution.
Dilution instructions
• Thawed vaccine must be diluted in its original vial with sodium chloride 9 mg/mL (0.9%)
solution for injection, using a 21 gauge or narrower needle and aseptic techniques. Volume
of sodium chloride 9 mg/mL (0.9%) required are noted below:
o
Yellow cap vials: 1.1 mL of sodium chloride 9 mg/mL
• Equalize vial pressure before removing the needle from the vial stopper by withdrawing air
into the empty diluent syringe. Volume of air required are noted below:
o
Yellow cap vials: 1.1 mL
of air
• Gently invert the diluted dispersion 10 times.
Do not shake.
• Check appearance of vaccine after dilution.
o
Yellow cap vials: : After mixing, the vaccine should present as a clear to slightly
opalescent dispersion with no particulates visible. Do not use the vaccine if
particulates or discoloration are present.
• After dilution, mark vial with appropriate date/time, store at 2 ºC to 30 ºC and use within
12 hours.
Do not re-freeze.
Preparation of individual doses
• Using aseptic technique, cleanse the vial stopper with a single-use antiseptic swab.
• Withdraw a single dose.
o
Yellow cap multidose vials (3 doses per vial):
Each dose must contain 0.3 mL of vaccine.
Standard syringes can be used.
• If the amount of vaccine remaining in the vial cannot provide a full dose, discard the vial
and any excess volume.
Any unused medicine or waste material should be disposed of in accordance with local
requirements.
7. MEDICINE SCHEDULE
Prescription Medicine.
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 40 of 41
8. SPONSOR
Pfizer New Zealand Limited
P O Box 3998
Auckland, New Zealand
Toll Free Number: 0800 736 363
9. DATE OF FIRST APPROVAL
Date of publication in the New Zealand Gazette of consent to distribute this medicine:
30 October 2025
10. DATE OF REVISION OF THE TEXT
24 November 2025
Comirnaty® is a registered trademark of BioNTech SE. Used under license.
Summary of Updates
Section
Update
N/A
New Data sheet
4.4
Addition of Study C4591024 data (immunocompromised)
4.8
Addition of AE data from Study C4591024
Addition of data from Study C4591054 SSA & SSB
Addition of data from Study C4591048 SSE
4.9
Inclusion of post-authorisation experience
5.1
Addition of Study C4591024 clinical data
Addition of data from Study C4591054 SSA & SSB
Addition of data from Study C4591048 SSE
Version: pfdclpyi11125
Supersedes: pfdclpyi11025
Page 41 of 41
Document 3
NEW ZEALAND DATA SHEET
1. PRODUCT NAME
Comirnaty® LP.8.1 COVID-19 mRNA vaccine , 10 micrograms/0.3 mL dose, suspension for
injection (light blue and dark blue caps), for age 5 to 11 years
2. QUALITATIVE AND QUANTITATIVE COMPOSITION
This is a single dose vial (light blue cap) or multidose vial (dark blue cap).
One light blue cap single dose vial (0.48 mL) contains 1 dose of 0.3 mL, see sections 4.2 and
6.6. One dose (0.3 mL) contains 10 micrograms of SARS-CoV-2 spike protein (mRNA)
LP.8.1, a COVID-19 mRNA Vaccine (embedded in lipid nanoparticles).
One dark blue cap multidose vial (2.25 mL) contains 6 doses of 0.3 mL, see sections 4.2 and
6.6. One dose (0.3 mL) contains 10 micrograms of SARS-CoV-2 spike protein (mRNA)
LP.8.1, a COVID-19 mRNA Vaccine (embedded in lipid nanoparticles).
SARS-CoV-2 spike protein (mRNA) LP.8.1 is a single-stranded, 5’-capped messenger RNA
(mRNA) produced using a cell-free
in vitro transcription from the corresponding DNA
templates, encoding the viral spike (S) protein of severe acute respiratory syndrome
coronavirus 2 (SARS-CoV-2) (LP.8.1).
For the full list of excipients, see Section 6.1 List of excipients.
3. PHARMACEUTICAL FORM
Comirnaty LP.8.1 suspension for injection (light blue and dark blue cap) is a clear to slightly
opalescent frozen suspension.
4. CLINICAL PARTICULARS
4.1 Therapeutic indications
Active immunisation to prevent coronavirus disease 2019 (COVID-19) caused by SARS-CoV-
2, in children aged 5 to 11 years.
The use of this vaccine should be in accordance with official recommendations.
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 1 of 37
4.2 Dose and method of administration
Dose
Strength & Age Group
Cap and Label Color
Volume of Each Dose
10 micrograms per dose
Light and dark blue
5 to 11 years
0.3 mL
Children 5 to 11 years of age
Comirnaty LP.8.1 10 micrograms/dose is administered intramuscularly as a single dose for
individuals 5 to 11 years of age, regardless of prior COVID-19 vaccination status.
For individuals who have previously been vaccinated with a COVID-19 vaccine, Comirnaty
LP.8.1 should be administered at least 3 months after the most recent dose of a COVID-19
vaccine.
Comirnaty LP.8.1 (Blue cap) should be used only for children 5 to 11 years of age.
Severely immunocompromised aged 5 years and older
Additional doses may be administered to individuals who are severely immunocompromised
in accordance with national recommendations (see section 4.4).
Paediatric population
There are paediatric formulations available for infants aged 6 months to 4 years of age. For
details, please refer to the data sheets for other fomulations. The safety and efficacy of the
vaccine in infants aged less than 6 months have not yet been established.
Elderly population
Refer to the Data Sheet for Comirnaty® LP.8.1, 30 micrograms/0.3 mL dose, suspension for
injection (light grey and dark grey caps) for individuals 12 years of age and older.
Method of administration
Comirnaty LP.8.1 should be administered intramuscularly. The preferred site of administration
is the deltoid muscle of the upper arm.
Do not inject the vaccine intravascularly, subcutaneously or intradermally.
Comirnaty LP.8.1 should not be mixed in the same syringe with any other vaccines or
medicinal products.
For precautions to be taken before administering the vaccine, see Section 4.4 Special warnings
and precautions for use. For instructions regarding thawing, handling and disposal of the
vaccine, see section 6.6.
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 2 of 37
Comirnaty LP.8.1 (Blue cap, Do not dilute)
Single dose vials
Single dose vials of Comirnaty LP.8.1 (light blue cap) contain 1 dose of 0.3 mL of vaccine
and do not require dilution.
- Withdraw a single 0.3 mL dose of Comirnaty LP.8.1
- Discard vial and any excess volume.
- Do not pool excess vaccine from multiple vials.
Multidose vials
Multidose vials of Comirnaty LP.8.1 (dark blue cap) contain 6 doses of 0.3 mL of vaccine and
do not require dilution.
In order to extract 6 doses from a multidose vial (dark blue cap), low dead-volume syringes
and/or needles should be used. The low dead-volume syringe and needle combination should
have a dead volume of no more than 35 microlitres. If standard syringes and needles are used,
there may not be sufficient volume to extract a sixth dose from a single vial. Irrespective of the
type of syringe and needle:
- Each dose must contain 0.3 mL of vaccine.
- If the amount of vaccine remaining in the vial cannot provide a full dose of 0.3 mL,
discard the vial and any excess volume.
- Do not pool excess vaccine from multiple vials.
For instructions on thawing, handling and dose preparation of Comirnaty LP.8.1 suspension
for injection, see Section 6.6 Special precautions for disposal and other handling.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of
excipients.
4.4 Special warnings and precautions for use
Traceability
In order to improve the traceability of biological medicinal products, the name and the batch
number of the administered product should be clearly recorded.
General recommendations
Hypersensitivity and anaphylaxis
Events of anaphylaxis have been reported. Appropriate medical treatment and supervision
should always be readily available in case of an anaphylactic reaction following the
administration of Comirnaty.
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 3 of 37
The individual should be kept under close observation for at least 15 minutes following
vaccination. A second dose of Comirnaty should not be given to those who have experienced
anaphylaxis to the first dose of Comirnaty.
Myocarditis and pericarditis
Very rare cases of myocarditis and pericarditis have been observed following vaccination with
Comirnaty. These cases have primarily occurred within 14 days following vaccination, more
often after the second vaccination, and more often, but not exclusively in younger men. There
have been reports in females. Based on accumulating data, the reporting rates of myocarditis
and pericarditis after primary series in children ages 5 to 11 years are lower than in ages 12 to
17 years. Rates of myocarditis and pericarditis in booster doses do not appear to be higher than
after the second dose in the primary series. The cases are generally mild and individuals tend
to recover within a short time following standard treatment and rest. Cases of myocarditis and
pericarditis following vaccination have rarely been associated with severe outcomes including
death.
Healthcare professionals should be alert to the signs and symptoms of myocarditis and
pericarditis, including atypical presentations. Vaccinees should be instructed to seek
immediate medical attention if they develop symptoms indicative of myocarditis or pericarditis
such as (acute and persisting) chest pain, shortness of breath, or palpitations following
vaccination. Non-specific symptoms of myocarditis and pericarditis also include fatigue,
nausea and vomiting, abdominal pain, dizziness or syncope, oedema and cough. Healthcare
professionals should consult guidance and/or specialists to diagnose and treat this condition.
Stress-related responses
Some individuals may have stress-related responses associated with the process of vaccination
itself. Stress-related responses are temporary and resolve on their own. They may include
dizziness, fainting, palpitations, increases in heart rate, alterations in blood pressure, feeling
short of breath, tingling sensations, sweating and/or anxiety. Individuals should be advised to
bring symptoms to the attention of the vaccination provider for evaluation and precautions
should be in place to avoid injury from fainting.
Concurrent il ness
Vaccination should be postponed in individuals suffering from acute severe febrile illness or
acute infection. The presence of a minor infection and/or low grade fever should not delay
vaccination.
Thrombocytopenia and coagulation disorders
As with other intramuscular injections, Comirnaty LP.8.1 should be given with caution in
individuals receiving anticoagulant ther apy or those with thrombocytopenia or any coagulation
disorder (such as haemophilia) because bleeding or bruising may occur following an
intramuscular administration in these individuals.
Immunocompromised individuals
Immunocompromised persons, including individuals receiving immunosuppressant therapy,
may have a diminished immune response to the vaccine.
Clinical data on safety and immunogenicity after administration of Comirnaty (tozinameran)
in immunocompromised participants are available in 37 participants 2 to 4 years old,
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 4 of 37
65 participants 5 to 11 years old, 15 participants 12 to 17 years old, and 7 participants 18 years
of age and older (see Sections 4.8 Undesirable effects and 5.1 Pharmacodynamic properties).
Duration of protection
The duration of protection afforded by Comirnaty is unknown as it is still being determined by
ongoing clinical trials.
Limitations of vaccine effectiveness
As with any vaccine, vaccination with Comirnaty may not protect all vaccine recipients.
Individuals may not be fully protected until 7 days after their second dose of Comirnaty.
Use in the elderly
Clinical studies of Comirnaty (tozinameran) include participants 65 years of age and older and
their data contributes to the overall assessment of safety and efficacy. See Section 5.1
Pharmacodynamic properties, Clinical trials, Efficacy against COVID-19.
Paediatric use
The safety and efficacy of Comirnaty in children aged less than 6 months of age have not yet
been established.
Effects on laboratory tests
No data available.
4.5 Interactions with other medicines and other forms of interactions
No interaction studies have been performed.
Concomitant administration of Comirnaty LP.8.1 (10 micrograms/dose) with other vaccines
has not been studied.
4.6 Fertility, pregnancy and lactation
Fertility
In a combined fertility and developmental toxicity study, female rats were intramuscularly
administered Comirnaty (tozinameran) prior to mating and during gestation (4 full human
doses of 30 micrograms each, spanning between pre-mating day 21 and gestation day 20).
SARS-CoV-2 neutralising antibodies were present in maternal animals from prior to mating to
the end of the study on postnatal day 21 as well as in fetuses and offspring. There were no
vaccine related effects on female fertility and pregnancy rate.
Pregnancy
No data are available yet regarding the use of Comirnaty LP.8.1 during pregnancy.
There are clinical study data from the use of Comirnaty (tozinameran) in 173 pregnant women
and no safety concerns were identified in the mother or their infant that were attributable to
maternal vaccination (see Section 4.8 Undesirable effects). Animal studies do not indicate
direct or indirect harmful effects with respect to pregnancy, embryo/fetal development,
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 5 of 37
parturition or post-natal development (see Section 4.6 Fertility, pregnancy and lactation,
Fertility).
Administration of Comirnaty LP.8.1 in pregnancy should only be considered when the
potential benefits outweigh any potential risks for the mother and fetus.
Lactation
No data are available yet regarding the use of Comirnaty LP.8.1 during breast-feeding. A
combined fertility and developmental toxicity study in rats did not show harmful effects on
offspring development before weaning (see Section 4.6 Fertility, pregnancy and lactation,
Fertility).
4.7 Effects on ability to drive and use machines
Comirnaty LP.8.1 has no, or negligible, influence on the ability to drive, cycle and use
machines. However, some of the effects mentioned under Section 4.8 Undesirable effects may
temporarily affect the ability to drive or use machines.
4.8 Undesirable effects
Summary of safety profile
The safety of Comirnaty (tozinameran) was evaluated in participants 5 years of age and older
in 3 clinical studies that included 24,675 participants (comprised of 22,026 participants 16
years of age and older, 1,131 adolescents 12 to 15 years of age from Study C4591001, and
3,109 children 5 to 11 years of age, 2,368 participants 2 to 4 years of age and 1,458 participants
6 to 23 months of age from Study C4591007) that have received at least one dose of Comirnaty
(tozinameran).
Additionally, 306 existing Phase 3 participants at 18 to 55 years of age received a booster dose
of Comirnaty (tozinameran) approximately 6 months after the second dose in the non-placebo-
controlled booster dose portion of Study C4591001. The overall safety profile for the booster
dose was similar to that seen after 2 doses.
In Study C4591031, a placebo-controlled booster study, 5,081 participants 16 years of age and
older were recruited from Study C4591001 to receive a booster dose of Comirnaty
(tozinameran) at least 6 months after the second dose. The overall safety profile for the booster
dose was similar to that seen after 2 doses.
In a subset of C4591007 Phase 2/3 participants, 401 participants 5 to 11 years of age received
a booster dose of Comirnaty at least 5 months after completing the primary series. The overall
safety profile for the booster dose was similar to that seen after the primary series.
In a subset of Study C4591054 (Substudy A, Phase 2/3), 412 participants 12 years of age and
older, who had received at least 3 doses of an mRNA COVID-19 vaccine, received a booster
dose of Comirnaty Omicron XBB.1.5. In another substudy of Study C4591054 (Substudy B,
Phase 2/3), 311 participants 12 years of age and older, who were COVID-19 vaccine-naïve,
received 1 dose of Comirnaty Omicron XBB.1.5. The safety profile of Comirnaty Omicron
XBB.1.5 was similar to the overall Comirnaty safety profile.
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 6 of 37
In a subset of C4591048 (Substudy E, Phase 2/3), 310 participants 5 to 11 years of age who
were COVID-19 vaccine-naïve, received 1 dose of Comirnaty Omicron XBB.1.5. The safety
profile of Comirnaty Omicron XBB.1.5 was similar to the overall Comirnaty safety profile.
Omicron-adapted Comirnaty
Participants 12 years of age and older – after a single dose in vaccine-naïve individuals
In a subset of C4591054 (Substudy B, Phase 2/3), 311 participants 12 years of age and older,
who were considered to be baseline SARS-CoV-2 positive and COVID-19 vaccine-naïve,
received 1 dose of Comirnaty Omicron XBB.1.5 (raxtozinameran). Participants had a median
follow-up time of 6.4 months up to a data cut-off date of 23 April 2024.
The safety profile of Comirnaty Omicron XBB.1.5 was similar to the overall Comirnaty safety
profile. The most frequent adverse reactions in participants were injection site pain (>50%),
fatigue (>30%), headache (>20%), chills (>10%), diarrhea (>10%), new or worsened muscle
pain (>10%), new or worsened joint pain (>10%), and swelling (>10%).
Participants 5 to 11 years of age – after a single dose in vaccine-naïve individuals
In a subset of C4591048 (Substudy E, Phase 2/3), 310 participants 5 to 11 years of age who
were COVID-19 vaccine-naïve, received 1 dose of Comirnaty Omicron XBB.1.5. Participants
had a median follow-up time of 6.4 months up to a data cut-off date of 1 November 2024.
The safety profile of Comirnaty Omicron XBB.1.5 was similar to the overall Comirnaty safety
profile. The most frequent adverse reactions in participants were pain at the injection site
(>40%), fatigue (>10%), headache (>10%), and new or worsened muscle pain (>10%).
Participants 12 years of age and older – after a booster dose
In a subset of C4591054 (Substudy A, Phase 2/3), 412 participants 12 years of age and older,
who had received at least 3 doses of an authorized mRNA COVID-19 vaccine, received a
booster dose of Comirnaty Omicron XBB.1.5.The safety profile of Comirnaty Omicron
XBB.1.5 was similar to the overall Comirnaty safety profile.
COMIRNATY (tozinameran)
Participants 16 years of age and older – after 2 doses
In Study C4591001, a total of 22,026 participants 16 years of age or older received at least 1
dose of Comirnaty (tozinameran) 30 micrograms and a total of 22,021 participants 16 years of
age or older received placebo (including 138 and 145 adolescents 16 and 17 years of age in the
Comirnaty (tozinameran) and placebo groups, respectively). A total of 20,519 participants 16
years of age or older received 2 doses of Comirnaty (tozinameran).
At the time of the analysis of Study C4591001 with a data cut-off of 13 March 2021 for the
placebo-controlled blinded follow-up period up to the participants’ unblinding dates, a total of
25,651 (58.2%) participants (13,031 Comirnaty (tozinameran) and 12,620 placebo) 16 years of
age and older were followed up for ≥4 months after the second dose. This included a total of
15,111 (7,704 Comirnaty (tozinameran) and 7,407 placebo) participants 16 to 55 years of age
and a total of 10,540 (5,327 Comirnaty (tozinameran) and 5,213 placebo) participants 56 years
and older.
The most frequent adverse reactions in participants 16 years of age and older that received 2
doses were injection site pain (>80%), fatigue (>60%), headache (>50%), myalgia (>40%),
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 7 of 37
chills (>30%), arthralgia (>20%), pyrexia and injection site swelling (>10%) and were usually
mild or moderate in intensity and resolved within a few days after vaccination. A slightly lower
frequency of reactogenicity events was associated with greater age.
The safety profile in 545 subjects receiving Comirnaty (tozinameran), that were seropositive
for SARS-CoV-2 at baseline, was similar to that seen in the general population.
Study C4591001 also included 200 participants with confirmed stable human
immunodeficiency virus (HIV) infection. The safety profile of the participants receiving
Comirnaty (tozinameran) (n=100) in the individuals with stable HIV infection was similar to
that seen in the general population.
Adolescents 12 to 15 years of age – after 2 doses
In an analysis of long term safety follow-up in Study C4591001, 2,260 adolescents
(1,131 Comirnaty (tozinameran) 30 micrograms; 1,129 placebo) were 12 to 15 years of age.
Of these, 1,559 adolescents (786 Comirnaty (tozinameran) and 773 placebo) have been
followed for ≥ 4 months after the second dose of Comirnaty (tozinameran). The safety
evaluation in Study C4591001 is ongoing.
The most frequent adverse reactions in adolescents 12 to 15 years of age that received 2 doses
were injection site pain (>90%), fatigue and headache (>70%), myalgia and chills (>40%),
arthralgia and pyrexia (>20%).
Children 5 to 11 years of age – after 2 doses
In an analysis of Study C4591007 Phase 2/3, 4,647 children (3,109 Comirnaty (tozinameran)
10 micrograms; 1,538 placebo) were 5 to 11 years of age. Of these, 2,206 (1,481 Comirnaty
(tozinameran) 10 micrograms and 725 placebo) children have been followed for >4 months
after the second dose in the placebo-controlled blinded follow-up period. The safety evaluation
in Study C4591007 is ongoing.
The most frequent adverse reactions in children 5 to 11 years of age that received 2 doses
included injection site pain (>80%), fatigue (>50%), headache (>30%), injection site redness
and swelling (≥20%), myalgia, chills and diarrhoea (>10%).
Participants 16 years of age and older – after booster dose
A subset from Study C4591001 Phase 2/3 participants of 306 adults 18 to 55 years of age who
completed the original Comirnaty (tozinameran) 2-dose course, received a booster dose of
Comirnaty (tozinameran) approximately 6 months (range of 4.8 to 8.0 months) after receiving
Dose 2. Of these, 301 participants have been followed for ≥4 months after the booster dose of
Comirnaty (tozinameran).
The most frequent adverse reactions in participants 18 to 55 years of age were injection site
pain (>80%), fatigue (>60%), headache (>40%), myalgia (>30%), chills and arthralgia (>20%).
In Study C4591031, a placebo-controlled booster study, participants 16 years of age and older
recruited from Study C4591001 received a booster dose of Comirnaty (tozinameran) (5,081
participants), or placebo (5,044 participants) at least 6 months after the second dose of
Comirnaty (tozinameran). Overall, participants who received a booster dose, had a median
follow-up time of 2.8 months (range 0.3 to 7.5 months) after the booster dose in the blinded
placebo-controlled follow-up period to the cut-off date (8 February 2022). Of these, 1281
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 8 of 37
participants (895 Comirnaty (tozinameran) and 386 placebo) have been followed for ≥ 4
months after the booster dose of Comirnaty (tozinameran).
Children 5 to 11 years of age – after booster dose
In a subset from C4591007, a total of 401 children 5 to 11 years of age received a booster dose
of Comirnaty (tozinameran) 10 micrograms at least 5 months (range of 5 to 9 months) after
completing the primary series. The analysis of the C4591007 Phase 2/3 subset is based on data
up to the cut-off date of 22 March 2022 (median follow-up time of 1.3 months).
The most frequent adverse reactions in participants 5 to 11 years of age were injection site pain
(>70%), fatigue (>40%), headache (>30%), myalgia, chills, injection site redness, and swelling
(>10%).
Tabulated list of adverse reactions from clinical studies and post-authorisation
experience
Adverse reactions observed during clinical studies are listed below according to the following
frequency categories:
Very common (≥ 1/10),
Common (≥ 1/100 to < 1/10),
Uncommon (≥ 1/1,000 to < 1/100),
Rare (≥ 1/10,000 to < 1/1,000),
Very rare (< 1/10,000),
Not known (cannot be estimated from the available data).
Table 1: Adverse reactions from Comirnaty (tozinameran) and Comirnaty Omicron
XBB.1.5 (raxtozinameran) clinical trials: Individuals 12 years of age and older
Rare
Not known
System Organ
Very
Common
Uncommon
(≥ 1/10,000
(cannot be
Class
common (≥ 1/100 to
(≥ 1/1,000 to
estimated from
(≥ 1/10)
< 1/10)
< 1/100)
to
< 1/1,000)
the available
data)
Blood and
Lymphadenopathya
lymphatic
system disorders
Metabolism and
Decreased appetite
nutrition
disorders
Psychiatric
Insomnia
disorders
Nervous system Headache
Lethargy
Acute
disorders
peripheral
facial
paralysisb
Gastrointestinal
Nausea;
disorders
Skin and
Hyperhidrosis;
subcutaneous
Night sweats
tissue disorders
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 9 of 37
Rare
Not known
System Organ
Very
Common
Uncommon
(≥ 1/10,000
(cannot be
Class
common (≥ 1/100 to
(≥ 1/1,000 to
estimated from
(≥ 1/10)
< 1/10)
< 1/100)
to
< 1/1,000)
the available
data)
Musculoskeletal Arthralgia;
and connective
Myalgia
tissue disorders
General
Injection
Injection
Asthenia; Malaise;
Facial swellingd
disorders and
site pain;
site redness
administration
Fatigue;
site conditions
Chills;
Pyrexiac;
Injection
site
swelling
a A higher frequency of lymphadenopathy (2.8% vs 0.4%) was observed in participants receiving a booster dose
in Study C4591031 compared to participants receiving 2 doses.
b Through the clinical trial safety follow-up period to 14 November 2020, acute peripheral facial paralysis (or
palsy) was reported by four participants in the Comirnaty (tozinameran) group. Onset was Day 37 after Dose 1
(participant did not receive Dose 2) and Days 3, 9, and 48 after Dose 2. No cases of acute peripheral facial paralysis
(or palsy) were reported in the placebo group.
c A higher frequency of pyrexia was observed after the second dose compared to the first dose. The preferred term
pyrexia is a cluster term covering also body temperature increased.
d Facial swelling in vaccine recipients with a history of injection of dermatological fillers
Table 2. Adverse Reactions from Comirnaty (tozinameran) and Comirnaty Omicron
XBB.1.5 (raxtozinameran) clinical trials: Individuals 5 to 11 Years of Age
(C4591007 22 May 2022 Data Cut-off Date, C4591048 SSE 10 October 2024
Study Completion Date)
Not known
Very
Common Uncommon
Rare
≥1/100 to ≥1/1,000 to ≥1/10,000 to
Very
(cannot be
System Organ Class Common
Rare
estimated
≥1/10
<1/10
<1/100
<1/1,000 <1/10,000 from the
(≥10%)
(≥1% to (≥0.1% to (≥0.01% to
<10%)
<1%)
<0.1%)
(<0.01%) available
data)
Blood and lymphatic
Lymphaden
system disorders
opathya
Immune system
Urticariab,c; Angioedemab,c
Anaphylaxisb
disorders
Pruritusb,c;
Rashb,c
Metabolism and
Decreased
nutrition disorders
appetite
Nervous system
Headache
disorders
Gastrointestinal
Diarrhoeab Vomitingb Nausea
disorders
Skin and subcutaneous
Night sweats
tissue disorders
Musculoskeletal and Myalgia
Arthralgia Pain in
connective tissue
extremity
disorders
(arm)b
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 10 of 37
General disorders and Injection Pyrexia
Malaise
administration site
site pain;
conditions
Fatigue;
Chills;
Injection
site
swelling;
Injection
site redness
a. A higher frequency of lymphadenopathy was observed in C4591007 (1.9% vs. 0.7%) in participants receiving a
booster dose compared to participants receiving 2 doses.
b. These adverse reactions were identified in the post-authorisation period. The following events were not
reported in participants 5 to 11 Years of Age in Study C4591007 but were reported in individuals ≥16 years of
age in Study C4591001: angioedema, lethargy, asthenia, hyperhidrosis, and night sweats.
c. The following events are categorised as hypersensitivity reactions: urticaria, pruritus, rash and angioedema
Special populations
Pregnant women and infants born to maternal participants – after 2 doses of Comirnaty
(tozinameran)
Study C4591015, a Phase 2/3, placebo-controlled study, evaluated Comirnaty (tozinameran)
or placebo administered in 2 doses, approximately 21 days apart, in pregnant women 18 years
of age and older, with the first dose given at 24 to 34 weeks gestation. A total of 346 pregnant
women received Comirnaty (tozinameran) (n=173) or placebo (n=173).
The most frequent adverse reactions in pregnant women who received any primary series dose
with Comirnaty (tozinameran) included injection site pain (>80%), fatigue (>60%),
headache (>50%), myalgia (>30%), chills, arthralgia, and injection site swelling (>10%).
The safety profile in pregnant women who received Comirnaty (tozinameran) was similar to
that of nonpregnant participants in other clinical studies, with no newly identified adverse
reactions.
In Study C4591015, safety in infants born to maternal participants who received Comirnaty
(tozinameran) (n=167) or placebo (n=168) was evaluated at birth and up to 6 months after birth.
No safety concerns were identified that were attributable to maternal vaccination with
Comirnaty (tozinameran).
Immunocompromised participants (adults and children)
In study C4591024, 37 participants 2 to 4 years old, 65 participants 5 to 11 years old, 15
participants 12 to 17 years old, and 7 participants 18 years of age and older from 5 different
immunocompromised disease subsets (immunomodulatory therapy, solid organ transplant,
stem cell transplant, non-small cell lung cancer (NSCLC)/chronic lymphocytic leukaemia
(CLL) and haemodialysis) received at least 1 and up to 4 doses of Comirnaty (tozinameran)
(Doses 1 and 2 were separated by 21 days, Doses 2 and 3 were separated by 28 days and Dose
4 was administered 3 to 6 months after Dose 3).
The safety profile in immunocompromised participants 2 years of age and older who received
Comirnaty (tozinameran) was similar to that in non-immunocompromised participants in other
clinical studies, with no newly identified adverse reactions.
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 11 of 37
Post-marketing experience
Although the events listed in Table 3 were not observed in the clinical trials, they are considered
adverse drug reactions for Comirnaty as they were reported in the post-marketing experience.
As these reactions were derived from spontaneous reports, the frequencies could not be
determined and are thus considered as not known.
Table 3: Adverse reactions from Comirnaty post marketing experience
System Organ Class
Adverse Drug Reaction
Immune system disorders
Anaphylaxis
Hypersensitivity reactions (e.g. rash, pruritis, urticaria, angioedema)
Cardiac disorders
Myocarditis
Pericarditis
Nervous system disorders
Dizziness
Gastrointestinal disorders
Diarrhoea
Vomiting
Musculoskeletal and connective Pain in extremity (arm)a
tissue disorders
General disorders and
Extensive swelling of vaccinated limb
administration site conditions
Reproductive system and breast Heavy menstrual bleedingb
disorders
a A higher frequency of pain in extremity (1.1% vs. 0.8%) was observed in participants receiving a booster dose in
Study C4591031 compared to participants receiving 2 doses.
b Most cases appear to be non-serious and temporary in nature.
Reporting suspected adverse effects
Reporting suspected adverse reactions after authorisation of the medicine is important. It allows
continued monitoring of the benefit/risk balance of the medicine. Healthcare professionals are
asked to report any suspected adverse reactions at
https://pophealth.my.site.com/carmreportnz/s/.
4.9 Overdose
In clinical trials, participants who received up to 2 times the recommended dose of Comirnaty
did not have an increase in reactogenicity or adverse reactions.
In post-authorisation experience, there have been reports of higher than recommended doses
of Comirnaty. In general, adverse events reported with overdoses have been similar to the
known adverse reaction profile of Comirnaty.
In the event of overdose, monitoring of vital functions and individualised symptomatic
treatment is recommended.
For risk assessment and advice on the management of overdose please contact the National
Poisons Centre on 0800 POISON (0800 764766).
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 12 of 37
5. PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: vaccines, other viral vaccines, ATC code: J07BN01.
Mechanism of action
The nucleoside-modified messenger RNA in Comirnaty is formulated in lipid nanoparticles,
which enable delivery of the non-replicating RNA into host cells to direct transient expression
of the SARS-CoV-2 spike (S) antigen. The mRNA codes for membrane-anchored, full-length
S with two point mutations within the central helix. Mutation of these two amino acids to
proline locks S in an antigenically preferred prefusion conformation. Comirnaty elicits both
neutralising antibody and cellular immune responses to the antigen, which may contribute to
protection against COVID-19.
Clinical efficacy and immunogenicity
Omicron-adapted Comirnaty
Immunogenicity in participants 12 years and older – after a single dose in vaccine-naïve
individuals
In a subset from C4591054, (Substudy B [Phase 2/3]), the evaluable immunogenicity
population of 302 vaccine-naïve participants 12 years of age and older who were considered to
be SARS−CoV-2 positive at baseline, received 1 dose of Comirnaty Omicron XBB.1.5, was
compared with participants in Substudy A [a subset from C4591054, (Phase 2/3)], who
received Comirnaty Omicron XBB.1.5 after at least 3 doses of an mRNA COVID-19 vaccine.
Neutralising titres against Omicron XBB.1.5 increased from baseline to 1 month after study
vaccination and were greater in participants receiving Comirnaty Omicron XBB.1.5 as a single
dose compared with participants who received Comirnaty Omicron XBB.1.5 after at least 3
doses of an mRNA COVID-19 vaccine. Noninferiority was met with respect to the geometric
mean ratio (GMR) of Omicron XBB.1.5-neutralising titres, and the difference in seroresponse
to the XBB.1.5 strain in Substudy B vaccine-naïve participants compared to the subset of
Substudy A (Table 4 and Table 5).
Table 4. Model-Based Geometric Mean Ratio – C4591054 Substudy B and Subset of
Substudy A – Evaluable Immunogenicity Population
Vaccine Group (as Assigned)
Group Comparison
Vaccine-Naïve
Vaccine-Experienced
Substudy B
Substudy A
Comirnaty
Comirnaty
Substudy B /
Omicron XBB.1.5
Omicron XBB.1.5
Substudy A
Sampling
30 mcg
30 mcg
Time
GMTc
GMTc
GMRd
Assaye
Pointa
nb
(95% CIc)
nb
(95% CIc)
(95% CId)
SARS-CoV-2
neutralisation
assay - Omicron
XBB.1.5 - NT50
4373.4
2915.7
1.93
(titre)e
1 month 299 (3757.1, 5090.9) 296
(2462.4, 3452.5)
(1.52, 2.44)f
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 13 of 37
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre;
LLOQ = lower limit of quantitation; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute respiratory
syndrome coronavirus 2.
a. Protocol-specified timing for blood sample collection.
b. n = Number of participants with valid and determinate assay results for the specified assay at both the
pre−vaccination time point and the given sampling time point.
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 ×
LLOQ.
d. GMRs and the corresponding 2-sided 95% CIs were calculated by exponentiating the difference in least
square means and the corresponding CIs based on a linear regression model with baseline assay results (log
scale), age, and vaccine group as covariates.
e. SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (Omicron subvariant
XBB.1.5).
f. Noninferiority is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67.
Table 5. Adjusted Difference in Percentages of Participants With Seroresponse –
C4591054 Substudy B and Subset of Substudy A – Evaluable Immunogenicity
Population
Vaccine Group (as Assigned)
Group Comparison
Vaccine-Naïve
Vaccine-Experienced
Substudy B
Substudy A
Comirnaty
Comirnaty
Omicron XBB.1.5
Omicron XBB.1.5
SARS-CoV-2
30 mcg
30 mcg
Adjusted Difference
Neutralisation Sampling
nc (%)
nc (%)
Differenc
Assayg
Time Pointa Nb
(95% CId)
Nb
(95% CId)
e %e
(95% CIf)
SARS-CoV-2
neutralisation
assay - Omicron
XBB.1.5 - NT50
253 (84.9)
218 (73.9)
(titre)g
1 month 298
(80.3, 88.8)
295
(68.5, 78.8)
7.31
(1.34, 13.28)h
Abbreviations: CI = confidence interval; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute respiratory
syndrome coronavirus 2.
a. Protocol-specified timing for blood sample collection.
b. N = number of participants with valid and determinate assay results for the specified assay at both the pre-
vaccination time point and the given sampling time point. These values are the denominators for the
percentage calculations.
c. n = Number of participants with a seroresponse for the given assay at the given sampling time point.
d. Exact 2-sided CI, based on the Clopper and Pearson method.
e. Difference in proportions, expressed as a percentage.
f. 2-Sided CI, based on the Miettinen and Nurminen method stratified by baseline neutralising titre category
(< median, ≥ median) and age group (< median, ≥ median). The median of baseline neutralising titres and
median age was calculated based on the pooled data in 2 comparator groups.
g. SARS-CoV-2 NT50 were determined using a validated 384-well assay platform Omicron subvariant
XBB.1.5).
h. Noninferiority is declared if the lower bound of the 2-sided 95% CI for the difference in percentages of
participants with seroresponse is >-10%.
Immunogenicity in participants 5 to 11 years of age – after a single dose in vaccine-naïve
individuals
In a subset from C4591048 (Substudy E [Phase 2/3]), the evaluable immunogenicity population
of 302 participants, who received a single 10 mcg dose of Comirnaty Omicron XBB.1.5 in
COVID-19 vaccine-naïve participants 5 to 11 years of age was compared to COVID-19
vaccine-experienced participants, 12 to 82 years of age, who received a single 30 mcg dose of
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 14 of 37
Comirnaty Omicron XBB.1.5 in C4591054 Substudy A. The majority of the participants were
considered to be SARS-CoV-2 positive at baseline (98.9% participants in C4591048 SSE,
99.3% participants in C4591054 Substudy A).
The primary immunobridging analyses compared the geometric mean titres (using a GMR) and
the seroresponse (defined as achieving at least 4-fold rise from baseline) rates in the
vaccine−naïve participants 5 to 11 years of age to COVID-19 vaccine-experienced participants
12 years of age and older. The immunobridging criteria were met for both the GMR and the
seroresponse rates (Table 6 and Table 7).
Table 6. Geometric Mean Ratio – C4591048 Substudy E to C4591054 Substudy A -
Participants at 1 Month After the Study Vaccination - Evaluable
Immunogenicity Population
C4591048 SSE
C4591054 SSA
5 to 11 Years of Age
12 Years of Age and older
Comirnaty Omicron XBB.1.5 Comirnaty Omicron XBB.1.5 C4591048 SSE /
SARS-CoV-2
10 mcg
30 mcg
C4591054 SSA
Neutralisation
GMTb
GMTb
GMRc
Assay
na
(95% CIb)
na
(95% CIb)
(95% CIc)
Omicron XBB.1.5
5930.5
4006.4
1.81
- NT50 (titre)d
285
(5283.8, 6656.4)
302
(3438.3, 4668.4)
(1.51, 2.16)e
Abbreviations: CI: confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre; LLOQ =
lower limit of quantitation; LS = least square; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute
respiratory syndrome coronavirus 2; SSA = Substudy A; SSE = Substudy E.
a. n = Number of participants with valid and determinate assay results for the specified assay at the given
sampling time point.
b. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 ×
LLOQ.
c. GMRs and 2-sided 95% CIs were calculated by exponentiating the difference of LS Means for the assay
(C4591048, 5 to 11 years of age – C4591054, 12 years of age and older) and the corresponding CIs based on
a linear regression model with baseline log-transformed neutralising titres, postbaseline infection status, and
vaccine group as covariates.
d. SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (Omicron subvariant
XBB.1.5).
e. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67 and
the point estimate of the GMR is ≥0.8.339
Table 7. Difference in Percentages of Participants With Seroresponse Between
C4591048 Substudy E and C4591054 Substudy A Participants at 1 Month
After the Study Vaccination - Evaluable Immunogenicity Population343
C4591048 SSE
C4591054 SSA
5 to 11 Years of Age
12 Years of Age and older
Comirnaty Omicron XBB.1.5 Comirnaty Omicron XBB.1.5
SARS-CoV-2
10 mcg
30 mcg
Difference
Neutralisation
nb (%)
nb (%)
Assay
Na
(95% CIc)
Na
(95% CIc)
%d
95% CIe
Omicron
XBB.1.5 - NT50
253 (88.8)
231 (77.0)
(3.91,
(titre)f
285
(84.5, 92.2)
300
(71.8, 81.6)
8.97
14.02)g
Abbreviations: CI: confidence interval; LLOQ = lower limit of quantitation; NT50 = 50% neutralising titre;
SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2; SSA = Substudy A; SSE = Substudy E.
Note: Seroresponse is defined as achieving a ≥4-fold rise from baseline. If the baseline measurement is below the
LLOQ, a post-vaccination assay result ≥4 × LLOQ is considered a seroresponse.
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 15 of 37
a. N = Number of participants with valid and determinate assay results for the specified assay both before
vaccination and at the given sampling time point. This value is the denominator for the percentage calculations.
b. n = Number of participants with seroresponse for the given assay at the given sampling time point.
c. Exact 2-sided 95% CI based on the Clopper and Pearson method.
d. Adjusted difference in proportions based on the Miettinen and Nurminen method stratified by baseline
neutralising titre category (<median, ≥median), expressed as a percentage (C4591048, 5 to 11 years of age –
C4591054, 12 years of age and older). The median of baseline neutralising titres was calculated based on the
pooled data in 2 comparator groups.
e. 2-sided 95% CI, based on the Miettinen and Nurminen method for the difference in proportions stratified by
baseline neutralising titre category (<median, ≥median), expressed as a percentage.
f. SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (Omicron subvariant
XBB.1.5).
g. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the adjusted difference in percentage
of participants with seroresponse is greater than -10.0%.
Immunogenicity in participants 12 years of age and older – after a booster dose
In a subset from C4591054 (Substudy A, Phase 2/3), the evaluable immunogenicity population
included 382 participants 12 years of age and older who had previously received at least 3 prior
doses of an authorized mRNA COVID-19 vaccine, with the most recent dose being an Omicron
BA.4/BA.5-adapted bivalent vaccine, received a booster dose of Comirnaty Omicron XBB.1.5.
At baseline, 78.8% of participants were considered to be positive for prior SARS−CoV−2
infection.
Compared to participants receiving Comirnaty Original/Omicron BA.4-5 (C4591044),
participants receiving Comirnaty Omicron XBB.1.5 (C4591054) had higher GMTs against
Omicron XBB.1.5 (2622.3 [CI: 2246.6, 3060.9] versus 601.0 [CI: 499.5, 723.1]) and against
Omicron BA.4/BA.5 (5105.1 [CI: 4483.4, 5813.0] versus 4146.0 [CI: 3512.6, 4893.5]) at 1
month after vaccination.
Seroresponse (NT50) was higher against Omicron XBB.1.5, and lower against Omicron
BA.4/BA.5 among participants who received Comirnaty Omicron XBB.1.5 at 1 month after
vaccination compared to the participants who Comirnaty Original/Omicron BA.4-5
(C4591044) with NT50 against Omicron XBB.1.5 of 73.9% (CI: 69.2%, 78.3%) versus
52.8% (CI: 45.6%, 59.9%), and NT50 against Omicron BA.4/BA.5 of 48.3% (CI: 43.2%,
53.4%) versus 63.0% (CI: 55.9%, 69.7%).
Comirnaty (tozinameran)
Study C4591001 is a multicentre, multinational, Phase 1/2/3 randomised, placebo-controlled,
observer-blind dose-finding, vaccine candidate selection and efficacy study in participants 12
years of age and older. Randomisation was stratified by age: 12 to 15 years of age, 16 to 55
years of age, or 56 years of age and older, with a minimum of 40% of participants in the ≥56-
year stratum. The study excluded participants who were immunocompromised and those who
had previous clinical or microbiological diagnosis of COVID-19. Participants with pre-existing
stable disease, defined as disease not requiring significant change in therapy or hospitalisation
for worsening disease during the 6 weeks before enrolment, were included as were participants
with known stable infection with HIV, hepatitis C virus (HCV) or hepatitis B virus (HBV).
Efficacy in participants 16 years of age and older – after 2 doses
In the Phase 2/3 portion of Study C4591001, based on data accrued through
14 November 2020, approximately 44,000 participants were randomised equally and were to
receive 2 doses of Comirnaty (tozinameran) or placebo. The efficacy analyses included
participants that received their second vaccination within 19 to 42 days after their first
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 16 of 37
vaccination. The majority (93.1%) of vaccine recipients received the second dose 19 days to
23 days after Dose 1. Participants are planned to be followed for up to 24 months after Dose 2,
for assessments of safety and efficacy against COVID-19. In the clinical study, participants
were required to observe a minimum interval of 14 days before and after administration of an
influenza vaccine in order to receive either placebo or Comirnaty (tozinameran). In the clinical
study, participants were required to observe a minimum interval of 60 days before or after
receipt of blood/plasma products or immunoglobulins through to conclusion of the study in
order to receive either placebo or Comirnaty (tozinameran).
The population for the analysis of the primary efficacy endpoint included 36,621 participants
12 years of age and older (18,242 in the Comirnaty (tozinameran) group and 18,379 in the
placebo group) who did not have evidence of prior infection with SARS-CoV-2 through 7 days
after the second dose. In addition, 134 participants were between the ages of 16 to 17 years of
age (66 in the Comirnaty (tozinameran) group and 68 in the placebo group) and 1616
participants 75 years of age and older (804 in the Comirnaty (tozinameran) group and 812 in
the placebo group).
At the time of the primary efficacy analysis, participants had been followed for symptomatic
COVID-19 for in total 2,214 person-years for the Comirnaty (tozinameran) group and in total
2,222 person-years for the placebo group.
There were no meaningful clinical differences in overall vaccine efficacy in participants who
were at risk of severe COVID-19 including those with 1 or more comorbidities that increase
the risk of severe COVID-19 (e.g. asthma, body mass index (BMI) ≥30 kg/m2, chronic
pulmonary disease, diabetes mellitus, hypertension).
Comirnaty (tozinameran) efficacy information is presented in Table 8.
Table 8: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2, by age
subgroup – participants without evidence of infection prior to 7 days after Dose 2 –
evaluable efficacy (7 days) population
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence of prior
SARS-CoV-2 infection*
Comirnaty
(tozinameran)
Placebo
Subgroup
Na = 18,198
Na = 18,325
Vaccine efficacy %
Cases n1b
Cases n1b
(95% CI)f
Surveillance timec (n2d) Surveillance timec (n2d)
All participantse
8
162
95.0
2.214 (17,411)
2.222 (17,511)
(90.0, 97.9)
16 to 64 years
7
143
95.1
1.706 (13,549)
1.710 (13,618)
(89.6, 98.1)
65 years and older
1
19
94.7
0.508 (3848)
0.511 (3880)
(66.7, 99.9)
65 to 74 years
1
14
92.9
0.406 (3074)
0.406 (3095)
(53.1, 99.8)
75 years and older
0
5
100.0
0.102 (774)
0.106 (785)
(-13.1, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 [*Case definition: (at least 1 of) fever, new or increased cough,
new or increased shortness of breath, chills, new or increased muscle pain, new loss of taste or smell, sore
throat, diarrhoea or vomiting.]
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 17 of 37
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence of prior
SARS-CoV-2 infection*
Comirnaty
(tozinameran)
Placebo
Subgroup
Na = 18,198
Na = 18,325
Vaccine efficacy %
Cases n1b
Cases n1b
(95% CI)f
Surveillance timec (n2d) Surveillance timec (n2d)
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the last dose) of
past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not
detected by nucleic acid amplification tests (NAAT) [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of
the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. No confirmed cases were identified in adolescents 12 to 15 years of age.
f. Two-sided confidence interval (CI) for vaccine efficacy (VE) is derived based on the Clopper and Pearson
method adjusted to the surveillance time. CI not adjusted for multiplicity.
In the second primary analysis, efficacy of Comirnaty (tozinameran) in preventing first
COVID-19 occurrence from 7 days after Dose 2 compared to placebo was 94.6% (95% credible
interval of 89.9% to 97.3%) in participants 16 years of age and older with or without evidence
of prior infection with SARS-CoV-2.
Additionally, subgroup analyses of the primary efficacy endpoint showed similar efficacy point
estimates across genders, ethnic groups, and participants with medical comorbidities associated
with high risk of severe COVID-19.
Updated efficacy analyses were performed with additional confirmed COVID-19 cases accrued
during blinded placebo-controlled follow-up through 13 March 2021, representing up to
6 months of follow-up after Dose 2 for participants in the efficacy population.
The updated vaccine efficacy information is presented in Table 9.
Table 9: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2, by age
subgroup – participants without evidence of infection prior to 7 days after Dose 2 –
evaluable efficacy (7 days) population during the placebo-controlled follow-up period
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
of prior SARS-CoV-2 infection*
Comirnaty
(tozinameran)
Placebo
Na=20,998
Na=21,096
Cases n1b
Cases n1b
Vaccine efficacy %
Subgroup
Surveil ance Timec (n2d) Surveil ance Timec (n2d)
(95% CIe)
All participantsf
77
850
91.3
6.247 (20,712)
6.003 (20,713)
(89.0, 93.2)
16 to 64 years
70
710
90.6
4.859 (15,519)
4.654 (15,515)
(87.9, 92.7)
65 years and older
7
124
94.5
1.233 (4192)
1.202 (4226)
(88.3, 97.8)
65 to 74 years
6
98
94.1
0.994 (3350)
0.966 (3379)
(86.6, 97.9)
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 18 of 37
75 years and older
1
26
96.2
0.239 (842)
0.237 (847)
(76.9, 99.9)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative
at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Two-sided confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method
adjusted to the surveillance time.
f. Included confirmed cases in participants 12 to 15 years of age: 0 in the Comirnaty (tozinameran) group (both
without and with or without evidence of prior SARS-CoV-2 infection); 16 and 18 in the placebo group
(without and with or without evidence of prior SARS-CoV-2 infection, respectively).
Efficacy against severe COVID-19 in participants 12 years of age or older – after 2 doses
As of 13 March 2021, vaccine efficacy against severe COVID-19 is presented only for
participants with or without prior SARS-CoV-2 infection (Table 10) as the COVID-19 case
counts in participants without prior SARS-CoV-2 infection were the same as those in
participants with or without prior SARS-CoV-2 infection in both the Comirnaty (tozinameran)
and placebo groups.
Table 10. Vaccine Efficacy – First Severe COVID-19 Occurrence in Participants
With or Without* Prior SARS-CoV-2 Infection Based on Food and Drug
Administration (FDA)† Definition After Dose 1 or From 7 Days After Dose 2 in the
Placebo-Controlled Follow-up
Comirnaty (tozinameran)
Placebo
Cases n1a
Cases n1a
Vaccine Efficacy %
Surveil ance Time (n2b) Surveil ance Time (n2b)
(95% CIc)
1
30
96.7
After Dose 1d
8.439e (22,505)
8.288e (22,435)
(80.3, 99.9)
1
21
95.3
7 days after Dose 2f
6.522g (21,649)
6.404g (21,730)
(70.9, 99.9)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at
least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or increased
shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat; diarrhoea;
vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative
at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
† Severe illness from COVID-19 as defined by FDA is confirmed COVID-19 and presence of at least 1 of the
following:
• Clinical signs at rest indicative of severe systemic illness (respiratory rate ≥30 breaths per minute, heart
rate ≥125 beats per minute, saturation of oxygen ≤93% on room air at sea level, or ratio of arterial
oxygen partial pressure to fractional inspired oxygen <300 mm Hg);
• Respiratory failure [defined as needing high-flow oxygen, noninvasive ventilation, mechanical
ventilation or extracorporeal membrane oxygenation (ECMO)];
• Evidence of shock (systolic blood pressure <90 mm Hg, diastolic blood pressure <60 mm Hg, or
requiring vasopressors);
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 19 of 37
• Significant acute renal, hepatic, or neurologic dysfunction;
• Admission to an Intensive Care Unit;
• Death.
a. n1 = Number of participants meeting the endpoint definition.
b. n2 = Number of participants at risk for the endpoint.
c. Two-side confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method
adjusted to the surveillance time.
d. Efficacy assessed based on the Dose 1 all available efficacy (modified intention-to-treat) population that
included all randomised participants who received at least 1 dose of study intervention.
e. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from Dose 1 to the end of the surveillance
period.
f. Efficacy assessed based on the evaluable efficacy (7 Days) population that included al eligible randomised
participants who receive all dose(s) of study intervention as randomised within the predefined window, have
no other important protocol deviations as determined by the clinician
g. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
Efficacy and immunogenicity in adolescents 12 to 15 years of age – after 2 doses
An analysis of Study C4591001 has been performed in adolescents 12 to 15 years of age up to
a data cutoff date of 13 March 2021.
The vaccine efficacy information in adolescents 12 to 15 years of age is presented in Table 11.
Table 11: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2 –
participants without evidence of infection and with or without evidence of infection prior
to 7 days after Dose 2 – adolescents 12 to 15 years of age evaluable efficacy (7 days)
population
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
without evidence of prior SARS-CoV-2 infection*
Comirnaty
(tozinameran)
Placebo
Na = 1005
Na = 978
Cases n1b
Cases n1b
Vaccine efficacy
Surveil ance timec (n2d) Surveil ance timec (n2d)
% (95% CIe)
Adolescents
0
16
12 to 15 years
0.154 (1001)
0.147 (972)
100.0 (75.3, 100.0)
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
with or without* evidence of prior SARS-CoV-2 infection
Comirnaty
(tozinameran)
Placebo
Na = 1119
Na = 1110
Cases n1b
Cases n1b
Vaccine efficacy
Surveil ance timec (n2d) Surveil ance timec (n2d)
% (95% CIe)
Adolescents
0
18
12 to 15 years
0.170 (1109)
0.163 (1094)
100.0 (78.1, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at
least 1 symptom consistent with COVID-19 [*Case definition: (at least 1 of) fever, new or increased cough, new
or increased shortness of breath, chills, new or increased muscle pain, new loss of taste or smell, sore throat,
diarrhoea or vomiting).
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the last dose) of
past SARS-CoV-2 infection (i.e, N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 20 of 37
detected by nucleic acid amplification tests (NAAT) [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all subjects within each group at
risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of subjects at risk for the endpoint.
e. Confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method adjusted
for surveillance time. CI not adjusted for multiplicity.
In Study C4591001 an analysis of SARS-CoV-2 neutralising titres in a randomly selected
subset of participants was performed to demonstrate non-inferior immune responses (within
1.5-fold) comparing adolescents 12 to 15 years of age to participants 16 to 25 years of age who
had no serological or virological evidence of past SARS-CoV-2 infection. The immune
response to Comirnaty (tozinameran) in adolescents 12 to 15 years of age (n = 190) was non-
inferior to the immune response in participants 16 to 25 years of age (n = 170), based on results
for SARS-CoV-2 neutralising titres at 1 month after Dose 2. The geometric mean titres (GMT)
ratio of the adolescents 12 to 15 years of age group to the participants 16 to 25 years of age
group was 1.76, with a 2-sided 95% CI of 1.47 to 2.10, meeting the 1.5-fold non-inferiority
criterion (the lower bound of the 2-sided 95% CI for the geometric mean ratio [GMR] >0.67),
which indicates a statistically greater response in the adolescents 12 to 15 years of age than that
of participants 16 to 25 years of age.
An updated efficacy analysis of Study C4591001 has been performed in approximately 2,260
adolescents 12 to 15 years of age evaluating confirmed COVID-19 cases accrued up to a data
cut-off date of 2 September 2021, representing up to 6 months of follow-up after Dose 2 for
participants in the efficacy population.
The updated vaccine efficacy information in adolescents 12 to 15 years of age is presented in
Table 12.
Table 12: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Dose 2:
Without Evidence of Infection and With or Without Evidence of Infection Prior to 7
Days After Dose 2 – Blinded Placebo-Controlled Follow-up Period, Adolescents 12 To
15 Years of Age Evaluable Efficacy (7 Days) Population
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
without evidence of prior SARS-CoV-2 infection*
Comirnaty
(tozinameran)
Placebo
Na=1057
Na=1030
Vaccine Efficacy
Cases n1b
Cases n1b
%
Surveil ance Timec (n2d) Surveil ance Timec (n2d)
(95% CIe)
Adolescents
0
28
100.0
12 to 15 years of age
0.343 (1043)
0.322 (1019)
(86.8, 100.0)
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of
age with or without evidence of prior SARS-CoV-2 infection
Comirnaty
(tozinameran)
Placebo
Na=1119
Na=1109
Vaccine Efficacy
Cases n1b
Cases n1b
%
Surveil ance Timec (n2d) Surveil ance Timec (n2d)
(95% CIe)
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 21 of 37
Adolescents
0
30
100.0
12 to 15 years of age
0.362 (1098)
0.345 (1088)
(87.5, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum]
negative at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had
negative NAAT (nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the
analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each
group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the
end of the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Two-sided confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson
method adjusted for surveillance time.
Efficacy in children 5 to 11 years of age – after 2 doses
An initial descriptive efficacy analysis of Study C4591007 has been performed in 1,968
children 5 to 11 years of age without evidence of infection prior to 7 days after Dose 2. This
analysis evaluated confirmed symptomatic COVID-19 cases accrued up to a data cut-off date
of 8 October 2021.
The initial descriptive vaccine efficacy results in children 5 to 11 years of age without evidence
of prior SARS-CoV-2 infection are presented in Table 13. None of the cases accrued met
criteria for severe COVID-19 or multisystem inflammatory syndrome in children (MIS-C). No
cases of COVID-19 were observed in either the vaccine group or the placebo group in
participants with evidence of prior SARS-CoV-2 infection.
Table 13: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Dose 2:
Without Evidence of Infection Prior to 7 Days After Dose 2 – Phase 2/3 – Children 5 To
11 Years of Age Evaluable Efficacy Population
First COVID-19 occurrence from 7 days after Dose 2 in children 5 to 11 years of age
without evidence of prior SARS-CoV-2 infection*
Comirnaty±
(tozinameran)
10 micrograms/dose
Placebo
Na=1305
Na=663
Vaccine Efficacy
Cases n1b
Cases n1b
%
Surveil ance Timec (n2d)
Surveil ance Timec (n2d)
(95% CI)
Children 5 to
3
16
90.7
11 years of age
0.322 (1273)
0.159 (637)
(67.7, 98.3)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum]
negative at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had
negative NAAT (nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the
analysis.
± Pfizer-BioNTech COVID-19 Vaccine (10 micrograms modRNA).
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 22 of 37
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each
group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the
end of the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
Prespecified hypothesis-driven efficacy analysis was performed with additional confirmed
COVID-19 cases accrued during blinded placebo-controlled follow-up, representing up to 6
months after Dose 2 in the efficacy population.
In the efficacy analysis of Study C4591007 in children 5 to 11 years of age without evidence
of prior infection, there were 10 cases out of 2,703 participants who received the vaccine and
42 cases out of 1,348 participants who received placebo. The point estimate for efficacy is
88.2% (95% CI: 76.2, 94.7). In participants with or without evidence of prior infection there
were 12 cases in the 3,018 who received vaccine and 42 cases in 1,511 participants who
received placebo. The point estimate for efficacy is 85.7% (95% CI: 72.4, 93.2).
Immunogenicity in children 5 to 11 years of age – after 2 doses
Study C4591007 is a Phase 1/2/3 study comprised of an open-label vaccine dose-finding
portion (Phase 1) and a multicentre, multinational, randomised, saline placebo-controlled,
observer-blind efficacy portion (Phase 2/3) that has enrolled participants 5 to 11 years of age.
In C4591007, an analysis of SARS-CoV-2 50% neutralising titres (NT50) 1 month after Dose
2 in a randomly selected subset of participants demonstrated effectiveness by immunobridging
of immune responses comparing children 5 to 11 years of age in the Phase 2/3 part of Study
C4591007 to participants 16 to 25 years of age in the Phase 2/3 part of Study C4591001 who
had no serological or virological evidence of past SARS-CoV-2 infection up to 1 month after
Dose 2, meeting the prespecified immunobridging criteria for both the geometric mean ratio
(GMR) and the seroresponse difference with seroresponse defined as achieving at least 4-fold
rise in SARS-CoV-2 NT50 from baseline (before Dose 1).
The ratio of the SARS-CoV-2 NT50 in children 5 to 11 years of age to that of young adults 16
to 25 years of age was 1.04 (2-sided 95% CI: 0.93, 1.18), as presented in Table 14.
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 23 of 37
Table 14: Summary of geometric mean ratio for 50% neutralising titre – Comparison
of children 5 to 11 years of age (Study C4591007) to participants 16 to 25 years of age
(Study C4591001) – participants without* evidence of infection up to 1 month after Dose
2 – evaluable immunogenicity population
Comirnaty (tozinameran)
10 microgram/dose 30 microgram/dose
5 to 11 years/
5 to 11 years
16 to 25 years
16 to 25 years
na=264
na=253
Met
Assay
Time
GMTc
GMTc
GMRd immunobridging
pointb
(95% CIc)
(95% CIc)
(95% CId)
objectivee
(Y/N)
SARS-CoV-2
neutralisation 1 month
1197.6
1146.5
1.04
assay - NT50 after
(1106.1, 1296.6)
(1045.5, 1257.2) (0.93, 1.18)
Y
(titre)f
Dose 2
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre;
LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test; NT50 = 50% neutralising titre;
SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
*Participants who had no serological or virological evidence (up to 1 month post-Dose 2 blood sample collection)
of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and 1 month after Dose
2, SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2, and negative NAAT (nasal swab) at
any unscheduled visit up to 1 month after Dose 2 blood collection) and had no medical history of COVID-
19 were included in the analysis.
a. n = Number of participants with valid and determinate assay results for the specified assay at the given
dose/sampling time point.
b. Protocol-specified timing for blood sample collection.
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
d. GMRs and 2-sided 95% CIs were calculated by exponentiating the mean difference of the logarithms of the
titres (Group 1[5 to 11 years of age] - Group 2 [16 to 25 years of age]) and the corresponding CI (based on
the Student t distribution).
e. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67 and
the point estimate of the GMR is ≥0.8.
f. SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation
Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus
neutralisation is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution
at which 50% of the virus is neutralised.
Among participants without prior evidence of SARS-CoV-2 infection up to 1 month after
Dose 2, 99.2% of children 5 to 11 years of age and 99.2% of participants 16 to 25 years of age
had a seroresponse from before vaccination to 1 month after Dose 2. The difference in
proportions of participants who had seroresponse between the 2 age groups (children – young
adult) was 0.0% (2-sided 95% CI: -2.0%, 2.2%) as presented in Table 15.
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 24 of 37
Table 15: Difference in percentages of participants with seroresponse – participants
without evidence of infection up to 1 month after Dose 2 – immunobridging subset –
Phase 2/3 – comparison of 5 to 11 years of age to Study C4591001 Phase 2/3 16 to 25 years
of age – evaluable immunogenicity population
Comirnaty (tozinameran)
10
30
5 to 11 years/
microgram/dose microgram/dose
16 to 25 years
5 to 11 years
16 to 25 years
Na=264
Na=253
Met
Assay
Time
nc (%)
nc (%)
Difference %e immunobridging
pointb
(95% CId)
(95% CId)
(95% CIf)
objectiveg
(Y/N)
SARS-CoV-2
neutralisation
1 month
262 (99.2)
251 (99.2)
0.0
assay – NT50 after
(97.3, 99.9)
(97.2, 99.9)
(-2.0, 2.2)
Y
(titre)h
Dose 2
Abbreviations: LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test;
N-binding = SARS-CoV-2 nucleoprotein–binding; NT50 = 50% neutralising titre 50; SARS-CoV-2 = severe acute
respiratory syndrome coronavirus 2.
Note: Seroresponse is defined as achieving a ≥4-fold rise from baseline (before Dose 1). If the baseline measurement
is below the LLOQ, a postvaccination assay result ≥4 × LLOQ is considered a seroresponse.
Note: Participants who had no serological or virological evidence (up to 1 month post-Dose 2 blood sample
collection) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and 1 month after
Dose 2, SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2, and negative NAAT (nasal swab) at
any unscheduled visit up to 1 month after Dose 2 blood collection) and had no medical history of COVID-19 were
included in the analysis.
a. N = number of participants with valid and determinate assay results both before vaccination and at 1 month after
Dose 2. These values are the denominators for the percentage calculations.
b. Protocol-specified timing for blood sample collection.
c. n = Number of participants with seroresponse for the given assay at the given dose/sampling time point.
d. Exact 2-sided CI based on the Clopper and Pearson method.
e. Difference in proportions, expressed as a percentage (Group 1 [5 to 11 years of age] – Group 2 [16 to 25 years
of age]).
f. 2-Sided CI, based on the Miettinen and Nurminen method for the difference in proportions, expressed as a
percentage.
g. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the difference in proportions is greater
than -10.0%.
h. SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation Assay.
The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus neutralisation is
read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution at which 50% of
the virus is neutralised.
Immunogenicity in participants 18 years of age and older – after booster dose
Effectiveness of a booster dose of Comirnaty (tozinameran) was based on an assessment of
50% neutralising titres (NT50) against SARS-CoV-2 (USA_WA1/2020). In Study C4591001,
analyses of NT50 1 month after the booster dose compared to 1 month after the primary series
in individuals 18 to 55 years of age who had no serological or virological evidence of past
SARS-CoV-2 infection up to 1 month after the booster vaccination demonstrated
noninferiority for both GMR and difference in seroresponse rates. Seroresponse for a
participant was defined as achieving a ≥4-fold rise in NT50 from baseline (before Dose 1),
These analyses are summarised in Table 16.
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 25 of 37
Table 16. SARS-CoV-2 neutralisation assay - NT50 (titre)† (SARS-CoV-2
USA_WA1/2020) – GMT and seroresponse rate comparison of 1 month after booster dose
to 1 month after primary series – participants 18 to 55 years of age without evidence of
infection up to 1 month after booster dose* – booster dose evaluable immunogenicity
population±
1 month after
booster dose/-
1 month after
Met
1 month after
1 month after
primary
noninferiority
booster dose
primary series
series
objective
n
(95% CI)
(95% CI)
(97.5% CI)
(Y/N)
Geometric mean
50% neutralising
2466.0b
755.7
b
3.26c
titre (GMTb)
212a (2202.6, 2760.8) (663.1, 861.2)
(2.76, 3.86)
Yd
Seroresponse rate
199f
190f
(%) for 50%
99.5%
95.0%
4.5%g
neutralising titre† 200e (97.2%, 100.0%) (91.0%, 97.6%) (1.0%, 7.9%
h)
Yi
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre;
LLOQ = lower limit of quantitation; N-binding = SARS-CoV-2 nucleoprotein-binding; NAAT = nucleic acid
amplification test; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute respiratory syndrome
coronavirus 2; Y/N = yes/no.
† SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation
Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus
neutralisation is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum
dilution at which 50% of the virus is neutralised.
* Participants who had no serological or virological evidence (up to 1 month after receipt of a booster dose
of Comirnaty) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative and
SARS-CoV-2 not detected by NAAT [nasal swab]) and had a negative NAAT (nasal swab) at any
unscheduled visit up to 1 month after the booster dose were included in the analysis.
± All eligible participants who had received 2 doses of Comirnaty (tozinameran) as initially randomised,
with Dose 2 received within the predefined window (within 19 to 42 days after Dose 1), received a booster
dose of Comirnaty (tozinameran), had at least 1 valid and determinate immunogenicity result after booster
dose from a blood collection within an appropriate window (within 28 to 42 days after the booster dose),
and had no other important protocol deviations as determined by the clinician.
a. n = Number of participants with valid and determinate assay results at both sampling time points within
specified window.
b. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
c. GMRs and 2-sided 97.5% CIs were calculated by exponentiating the mean differences in the logarithms of
the assay and the corresponding CIs (based on the Student t distribution).
d. Noninferiority is declared if the lower bound of the 2-sided 97.5% CI for the GMR is > 0.67 and the point
estimate of the GMR is ≥ 0.80.
e. n = Number of participants with valid and determinate assay results for the specified assay at baseline,
1 month after Dose 2 and 1 month after the booster dose within specified window. These values are the
denominators for the percentage calculations.
f. Number of participants with seroresponse for the given assay at the given dose/sampling time point. Exact
2-sided CI based on the Clopper and Pearson method.
g. Difference in proportions, expressed as a percentage (1 month after booster dose – 1 month after Dose 2).
h. Adjusted Wald 2-sided CI for the difference in proportions, expressed as a percentage.
i. Noninferiority is declared if the lower bound of the 2-sided 97.5% CI for the percentage difference is
> -10%.
Relative vaccine efficacy in participants 16 years of age and older – after booster dose
An interim efficacy analysis of Study C4591031, a placebo-controlled booster study, was
performed in approximately 10,000 participants 16 years of age and older who were recruited
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 26 of 37
from Study C4591001, evaluated confirmed COVID-19 cases accrued from at least 7 days after
booster vaccination up to a data cut-off date of 8 February 2022 (a period when Delta and then
Omicron was the predominant variant), which represents a median of 2.8 months (range 0.3 to
7.5 months) post-booster follow-up. Vaccine efficacy of the Comirnaty (tozinameran) booster
dose after the primary series relative to the placebo booster group who only received the
primary series dose was assessed. The relative vaccine efficacy information for participants
16 years of age and older is presented in Table 17.
Table 17: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Booster
Vaccination – Participants 16 Years of Age and Older Without Evidence of Infection
and Participants With or Without Evidence of Infection Prior to 7 Days After Booster
Vaccination – Evaluable Efficacy Population
First COVID-19 occurrence from 7 days after booster dose in participants without evidence
of prior SARS-CoV-2 infection*
Comirnaty
(tozinameran)
Placebo
Na=4689
Na=4664
Relative Vaccine
Cases n1b
Cases n1b
Efficacye %
Surveil ance Timec (n2d) Surveil ance Timec (n2d)
(95% CIf)
First COVID-19
occurrence from
7 days after booster
63
148
63.9
vaccination
1.098 (4639)
0.932 (4601)
(51.1, 73.5)
First COVID-19 occurrence from 7 days after booster dose in participants with or without
evidence of prior SARS-CoV-2 infection
Comirnaty
(tozinameran)
Placebo
Na=4977
Na=4942
Relative Vaccine
Cases n1b
Cases n1b
Efficacye %
Surveil ance Timec (n2d) Surveil ance Timec (n2d)
(95% CIf)
First COVID-19
occurrence from
7 days after booster
67
150
62.4
vaccination
1.173 (4903)
0.989 (4846)
(49.5, 72.2)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the booster
vaccination) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and
SARS-CoV-2 not detected by NAAT [nasal swab] at Visit 1, and had a negative NAAT [nasal swab] at
any unscheduled visit prior to 7 days after booster vaccination) were included in the analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each
group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after the booster
vaccination to the end of the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Relative vaccine efficacy of the Comirnaty (tozinameran) booster group relative to the placebo group
(non-booster).
f. Two-sided confidence interval (CI) for relative vaccine efficacy is derived based on the Clopper and
Pearson method adjusted for surveillance time.
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 27 of 37
Immunogenicity in children 5 to 11 years of age – after booster dose
Effectiveness of a booster dose of Comirnaty (tozinameran) was based on an assessment of
NT50 against the reference strain of SARS-CoV-2 (USA_WA1/2020). Analyses of NT50
1 month after the booster dose compared to before the booster dose demonstrated a substantial
increase in GMTs in individuals 5 to 11 years of age who had no serological or virological
evidence of past SARS-CoV-2 infection up to 1 month after the booster dose. This analysis is
summarised in Table 18.
Table 18: Summary of Geometric Mean Titres – NT50 – Participants Without
Evidence of Infection – Phase 2/3 – Immunogenicity Set – 5 to 11 Years of Age – Evaluable
Immunogenicity Population
Comirnaty (tozinameran) 10 mcg/Dose
3-Dose Set
2-Dose Set
Total
Dose/
Sampling
GMTc
GMTc
GMTc
Assay
Time Pointa
nb
(95% CIc)
nb
(95% CIc) nb
(95% CIc)
20.5
20.5
20.5
1 month Prevax 79
(20.5, 20.5)
67 (20.5, 20.5) 146 (20.5, 20.5)
SARS-CoV-2 1 month after Dose
1659.4
1110.7
1253.9
neutralisation
2
29 (1385.1, 1988.0) 67 (965.3, 1278.1) 96 (1116.0, 1408.9)
assay - NT50
271.0
271.0
(titre)
3 months Prevax 67 (229.1, 320.6) -
-
67 (229.1, 320.6)
1 month after Dose
2720.9
2720.9
3
67 (2280.1, 3247.0) -
-
67 (2280.1, 3247.0)
Abbreviations: CI = confidence interval; GMT = geometric mean titre; LLOQ = lower limit of quantitation;
NAAT = nucleic acid amplification test; N-binding = SARS-CoV-2 nucleoprotein–binding; NT50 = 50%
neutralising titre; Prevax = before vaccination; SARS-CoV-2 = severe acute respiratory syndrome coronavirus
2.
Note: Three-dose immunogenicity set included the first 130 participants who received Dose 3 and completed
1-month post–Dose 3 visit prior to March 15, 2022. Among those, 30 had blood sample collection at 1-month
post-Dose 2. Two-dose immunogenicity set included an extra 67 participants randomly selected from previous
Dose-2 evaluable immunogenicity population and without evidence of infection up to 1-month post–Dose 2
subset used for 2-dose immunobridging analysis.
Note: Participants included in this analysis had no serological or virological evidence of past SARS-CoV-2
infection up to the 1-month post–Dose 2 (for 1-month post–Dose 2 time point) or 1-month post–Dose 3 (for pre–
Dose 3 and 1-month post–Dose 3 time point) study blood sample collection. Having no evidence of past SARS-
CoV-2 infection up to 1-month post–Dose 2 was defined as having a negative N-binding antibody (serum) result
at the Dose 1 and 1-month post–Dose 2 study visits; a negative NAAT (nasal swab) result at the Dose 1 and
Dose 2 study visits and any unscheduled visit prior to the 1-month post–Dose 2 blood sample collection; and no
medical history of COVID-19. Having no evidence of past SARS-CoV-2 infection up to 1-month post-Dose 3
was defined as having a negative N-binding antibody (serum) result at the Dose 1, 1-month post–Dose 2 (if
available), Dose 3, and 1-month post–Dose 3 study visits; a negative NAAT (nasal swab) result at the Dose 1,
Dose 2, and Dose 3 study visits and any unscheduled visit prior to the 1-month post–Dose 3 blood sample
collection; and no medical history of COVID-19.
a. Protocol-specified timing for blood sample collection.
b. n = Number of participants with valid and determinate assay results for the specified assay at the given
dose/sampling time point.
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 ×
LLOQ.
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 28 of 37
Immunogenicity in children 5 to 11 years of age on the Omicron variant (B1.1.529) –
after booster dose
The neutralising GMTs against both the Omicron variant (B1.1.529) and reference strain were
substantially increased after booster vaccination compared with after the 2-dose primary series.
At 1-month post-Dose 2, the observed neutralising GMTs for the Omicron variant (B1.1.529)
and reference strain were 27.6 and 323.8, respectively. At 1-month post-Dose 3, the observed
neutralising GMTs for the Omicron variant (B1.1.529) and reference strain were 614.4 and
1702.8, respectively (see Table 19).
For the Omicron variant (B1.1.529), neutralising titres after booster vaccination (1-month post-
Dose 3) increased 22-fold over those after the 2-dose primary series (1-month post-Dose 2).
For the reference strain, the increase after the booster relative to the primary series was 5.3-
fold.
Table 19: Summary of Geometric Mean Titres – Omicron-Neutralisation Subset –
Participants Without Evidence of Infection – Phase 2/3 – Immunogenicity Set – 5 to 11
Years of Age – Evaluable Immunogenicity Population
Comirnaty (tozinameran)
10 mcg/Dose
Vaccine Group (as Randomised)
GMTc
Assay
Time Pointb
nb
(95% CIc)
SARS-COV-2 FFRNT-
27.6
B.1.1.529 strain
1 month after Dose 2
29
(22.1, 34.5)
(Omicron) - NT50
614.4
(titre)
1 month after Dose 3
17
(410.7, 919.2)
SARS-CoV-2 FFRNT-
323.8
reference strain - NT50
1 month after Dose 2
29
(267.5, 392.1)
(titre)
1702.8
1 month after Dose 3
17
(1282.6, 2260.7)
Abbreviations: CI = confidence interval; FFRNT = fluorescence focus reduction neutralisation test;
GMT = geometric mean titre; LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test;
N-binding = SARS-CoV-2 nucleoprotein–binding; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute
respiratory syndrome coronavirus 2.
Note: Participants included in this analysis had no serological or virological evidence of past SARS-CoV-2
infection up to the 1-month post–Dose 2 (for 1-month post–Dose 2 time point) or 1-month post–Dose 3 (for
1-month post–Dose 3 time point) study blood sample collection. Having no evidence of past SARS-CoV-2
infection up to 1-month post–Dose 2 was defined as having a negative N-binding antibody (serum) result at
the Dose 1 and 1-month post–Dose 2 study visits; a negative NAAT (nasal swab) result at the Dose 1 and Dose
2 study visits and any unscheduled visit prior to the 1-month post–Dose 2 blood sample collection; and no
medical history of COVID-19. Having no evidence of past SARS-CoV-2 infection up to 1-month post–Dose 3
was defined as having a negative N-binding antibody (serum) result at the Dose 1, 1-month post–Dose 2 (if
available), Dose 3, and 1-month post–Dose 3 study visits; a negative NAAT (nasal swab) result at the Dose 1,
Dose 2, and Dose 3 study visits and any unscheduled visit prior to the 1-month post–Dose 3 blood sample
collection; and no medical history of COVID-19.
a. Protocol-specified timing for blood sample collection.
b. n = Number of participants with valid and determinate assay results for the specified assays at the given
dose/sampling time point.
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 29 of 37
Immunogenicity in pregnant women and infants born to maternal participants – after 2
doses with Comirnaty (tozinameran)
Study C4591015 was a Phase 2/3 multinational, placebo-controlled, observer-blind study that
enrolled pregnant women 18 years of age and older to receive 2 doses of Comirnaty
(tozinameran) (n=173) or placebo (n=173). Pregnant women received Dose 1 of Comirnaty
(tozinameran) at 24 to 34 weeks gestation and the majority (90.2%) received the second dose
19 to 23 days after Dose 1.
Descriptive immunogenicity analysis was performed in pregnant women receiving Comirnaty
(tozinameran) in Study C4591015 compared to a comparator subset of nonpregnant women
from Study C4591001 evaluating the ratio of the neutralising GMT (GMR) 1 month after Dose
2.
The evaluable immunogenicity population who received Comirnaty (tozinameran) in the
maternal participants group in Study C4591015 (n=111) and in nonpregnant participants in
Study C4591001 (n=114) comprised of 69.4% vs. 82.5% White, 27.0% vs. 5.3% Black or
African American, 1.8% vs. 6.1% Asian, 0 vs 4.4% multiracial participants, 37.8% vs 34.2%
Hispanic/Latino, 37.8% vs 3.5% had a positive baseline SARS-CoV-2 status, and 38.7% vs
27.2% were obese [BMI ≥30 kg/m2 (pre-pregnancy weight in participants in Study
C4591015)], respectively. In maternal participants group in Study C4591015 and in
nonpregnant participants in Study C4591001 who received Comirnaty (tozinameran), the
median age was 30 years (range 18 to 44 years of age) in both groups.
The immunogenicity results after 2 doses of Comirnaty (tozinameran) in pregnant women
18 years of age and older are presented in Table 20.
Table 20. Geometric Mean Ratios – Participants Without* or With or Without Evidence
of Infection up to 1 Month After Dose 2 – Maternal Participants (Study
C4591015) and Nonpregnant Female Participants (Study C4591001) –
Evaluable Immunogenicity Population
Participants Without Evidence of Infection*
Comirnaty (tozinameran)
Study C4591015
Study C4591001
Pregnant/
Pregnant Women
Nonpregnant Women Nonpregnant
Dose/
Sampling
GMTd
GMTd
GMRe
Assay
Time Pointb nc
(95% CId)
nc
(95% CId)
(95% CI)e
SARS-CoV-2
neutralisation
assay - NT50
1109.2
1663.7
0.67
(titre)a
2/1 month
58
(849.2, 1448.9) 107 (1411.5, 1960.8)
(0.50, 0.90)
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 30 of 37
Participants With or Without Evidence of Infection
Comirnaty (tozinameran)
Study C4591015
Study C4591001
Pregnant/
Pregnant Women
Nonpregnant Women Nonpregnant
Dose/
Sampling
GMTg
GMTg
GMRh
Assay
Time Pointb nf
(95% CIg)
nf
(95% CIg)
(95% CI)h
SARS-CoV-2
neutralisation
assay - NT50
1900.0
2005.7
0.95
(titre)a
2/1 month
99 (1518.2, 2377.7) 113 (1627.0, 2472.6)
(0.69, 1.30)
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre; LLOQ = lower limit
of quantitation; LS = least square; N-binding = SARS-CoV-2 nucleoprotein–binding; NAAT = nucleic acid amplification
test; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
Note: Participants from Study 2 are a selected subset of age matched nonpregnant female Phase 3 participants.
* Participants who had no serological or virological evidence (prior to the 1 month after Dose 2 blood sample collection)
of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Dose 1 and 1 month after Dose 2 and no
positive result between visits, negative NAAT [nasal swab] at Dose 1, Dose 2, and any unscheduled visit prior to the 1
month after Dose 2 blood sample collection) were included in the analysis.
a. SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (original strain [USA-WA1/2020,
isolated in January 2020]).
b. Protocol-specified timing for blood sample collection.
c. n = Number of participants with valid and determinate assay results for the specified assay at the given dose/sampling
time point.
d. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the corresponding
CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 × LLOQ.
e. GMR and 2-sided 95% CIs were calculated by exponentiating the mean difference of the logarithms of the assay and
the corresponding CIs (based on the Student t distribution).
f. n = Number of participants with valid and determinate assay results for the specified assay at both baseline and the
given dose/sampling time point.
g. GMTs and 2-sided CIs were calculated by exponentiating the LS means and the corresponding CIs based on analysis of
log-transformed NT50 titres using a regression model with group, age at Dose 1 in years (continuous), and baseline
log-transformed NT50 titres.
h. GMR (ratio of GMTs of pregnant women to nonpregnant women) and 2-sided CIs were calculated by exponentiating
the difference of LS means and the corresponding CIs based on the same regression model as above.
Among participants without prior evidence of SARS-CoV-2 infection up to 1 month after
Dose 2 (evaluable immunogenicity population), the ratio of the neutralising GMTs (GMR) in
Study C4591015 maternal participants in the BNT162b2 (30 μg) group to that of Study
C4591001 nonpregnant females who received BNT162b2 30 μg was 0.67 (95% CI: 0.50,
0.90).
For participants with or without prior evidence of SARS-CoV-2 infection up to 1 month after
Dose 2 (evaluable immunogenicity population), the model-adjusted ratio of the neutralising
GMTs (adjusted GMR) in Study C4591015 maternal participants in the BNT162b2 (30 μg)
group to that of Study C4591001 nonpregnant females who received BNT162b2 30 μg was
0.95 (95% CI: 0.69, 1.30). The model-adjusted GMT and GMR were calculated based on a
regression model adjusting for age and baseline neutralising titres.
In an additional descriptive immunogenicity analysis, infants born to maternal participants who
received COMIRNATY (tozinameran) had higher geometric mean concentrations (GMCs) of
full length S-binding immunoglobulin G (IgG) concentrations at birth and at 6 months after
delivery [5576.4 (95% CI: 4246.2, 7323.2); n=91 and 311.1 (95% CI: 235.8, 410.5); n=83],
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 31 of 37
respectively, compared to infants born to maternal participants from the placebo group [19.4
(95% CI: 10.2, 37.0); n=92 and 22.0 (95% CI: 11.4, 42.7); n=69].
Immunogenicity in immunocompromised participants (adults and children)
Study C4591024 is a Phase 2b, open-label study (n=124) that enrolled immunocompromised
participants 2 to 17 years of age receiving immunomodulator therapy or who have undergone
solid organ transplant (within the previous 3 months) and are on immunosuppression or who
have undergone bone marrow or stem cell transplant at least 6 months prior to enrollment.
Study C4591024 also enrolled immunocompromised participants 18 years of age and older
treated for NSCLC or CLL, receiving hemodialysis for secondary to end-stage renal disease,
or receiving immunomodulator therapy for an autoimmune inflammatory disorder. Study
participants did not have a past clinical or microbiological diagnosis of COVID-19. Participants
received 4 age-appropriate doses of Comirnaty (tozinameran) (3 micrograms, 10 micrograms,
or 30 micrograms); the first 2 doses separated by 21 days, with the third dose occurring 28 days
after the second dose, followed by a fourth dose, 3 to 6 months after Dose 3.
The immunogenicity results pre-vaccination and after 3 and 4 doses of Comirnaty
(tozinameran) in immunocompromised participants 2 years of age and older are presented in
Table 21.
Table 21. Summary of Geometric Mean Titres – Participants With or Without
Evidence of Infection by Age Group – All-Available Immunogenicity
Population
Comirnaty (tozinameran)
3 micrograms
10 micrograms
30 micrograms 30 micrograms
Age Group:
Age Group:
Age Group:
Age Group:
2 to 4 Years
5 to 11 Years
12 to 17 Years
≥
18 Years
Dose/
Sampling
GMTc
GMTc
GMTc
GMTc
Assay
Time Pointb nc
(95% CId)
nc (95% CId) nc
(95% CId) nc (95% CId)
SARS-CoV-2
44.8
44.5
54.2
82.2
neutralisation
1/Prevax
32
(42.2, 47.7)
62 (42.5, 46.5) 14 (33.7, 87.0) 6 (16.0, 422.5)
assay –
1566.5
2940.6
787.1
reference strain
942.3
(1019.9,
(1175.5,
(66.5,
– NT50 (titre)a 3/1 Month 32 (537.1, 1653.4) 60
2405.9)
14
7356.0)
6
9321.5)
922.2
3284.5
606.2
487.8
(586.7,
(1609.8,
(5.3,
4/Pre-Dose 4 29
(269.0, 884.9) 57
1449.3)
11
6701.3)
3
68756.0)
6463.4
13457.1
1031.3
3447.0
(4319.7,
(5270.1,
(56.9,
4/1 Month 26 (1851.0, 6419.2) 50
9670.9)
9
34362.4)
4
18681.7)
2382.3
5776.1
1605.6
1296.7
(1554.3,
(2801.4,
(28.5,
4/6 Months 25 (674.2, 2494.0) 49
3651.2)
8
11909.2)
3
90614.9)
Abbreviations: CI = confidence interval; GMT = geometric mean titre; LLOQ = lower limit of quantitation; NT50 = 50%
neutralising titre; Prevax = before vaccination; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
a. SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (original strain [USA-WA1/2020, isolated
in January 2020]).
b Protocol-specified timing for blood sample collection.
c. n = Number of participants with valid and determinate assay results for the specified assay at the given dose/sampling time
point.
d. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the corresponding CIs
(based on the Student t distribution). Assay results below the LLOQ were set to 0.5 × LLOQ.
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 32 of 37
Analysis of immunogenicity data at 1 month after Dose 3 (32 participants 2 to 4 years of age,
60 participants 5 to 11 years of age, 14 participants 12 to 17 years of age, and 6 participants
18 years of age and older) and 1 month after Dose 4 (26 participants 2 to 4 years of age, 50
participants 5 to 11 years of age, 9 participants 12 to 17 years of age, and 4 participants 18
years of age and older) in the all available immunogenicity population with or without evidence
of prior infection demonstrated a vaccine-elicited immune response.
GMTs were observed to be substantially higher at 1 month after Dose 3 and further increased
at 1 month after Dose 4 and remained high at 6 months after Dose 4 compared to levels
observed before study vaccination across age groups and disease subsets.
5.2 Pharmacokinetic properties
Not applicable.
5.3 Preclinical safety data
Genotoxicity/Carcinogenicity
Neither genotoxicity nor carcinogenicity studies were performed. The components of
Comirnaty (lipids and mRNA) are not expected to have genotoxic potential.
6. PHARMACEUTICAL PARTICULARS
6.1 List of excipients
((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC)
Cholesterol
Trometamol
Trometamol hydrochloride
Sucrose
Water for injections
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those
mentioned in Section 6.6 Special precautions for disposal and other handling.
6.3 Shelf life
Unopened vial
Frozen vial
18 months when stored at -90°C to -60°C.
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 33 of 37
The vaccine will be received frozen at -90°C to -60°C. Frozen vaccine can be stored either at
-90°C to -60°C or 2°C to 8°C upon receipt.
For thawing instructions of the frozen vials, see Section 6.6 Special precautions for disposal
and other handling.
Thawed vial
If the vaccine is received at 2°C to 8°C it should be stored at 2°C to 8°C. Once removed from
frozen storage, the unopened vial may be stored refrigerated at 2°C to 8°C for a single period
of up to 10 weeks within the 18 month shelf life.
Upon moving the product to 2°C to 8°C storage, the updated expiry date must be written on
the outer carton and the vaccine should be used or discarded by the updated expiry date. The
original expiry date should be crossed out.
Check that the expiry date on the outer carton has been updated to reflect the refrigerated expiry
date and that the original expiry date has been crossed out.
Prior to use, the unopened vials can be stored for up to 12 hours at temperatures between
8ºC to 30ºC.
Thawed vials can be handled in room light conditions.
Once thawed the vaccine should not be re-frozen.
Opened vial (Blue caps)
Chemical and physical in-use stability has been demonstrated for 12 hours at 2ºC to 30ºC.
From a microbiological point of view, unless the method of opening precludes the risks of
microbial contamination, the product should be used immediately after the first puncture. If not
used immediately, in-use storage times and conditions are the responsibility of the user.
6.4 Special precautions for storage
Check that the expiry date has been updated to reflect the refrigerated EXP date and that the
original expiry date has been crossed out.
Store in the original package to protect from light. During storage, minimise exposure to room
light, and avoid exposure to direct sunlight and ultraviolet light.
For detailed instructions see Section 6.6 Special precautions for disposal and other handling.
Once thawed, the vaccine cannot be re-frozen.
Thawed vials can be handled in room light conditions.
For storage conditions after thawing and dilution of the medicinal product, see Section 6.3
Shelf life.
For additional advice on storing Comirnaty LP.8.1, contact Pfizer New Zealand on 0800 736
363.
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 34 of 37
6.5 Nature and contents of container
Comirnaty LP.8.1 (Light blue cap) 0.48 mL fill volume, 2 mL clear vial (Type I glass) with a
stopper (synthetic bromobutyl rubber) and a Light Blue flip-off plastic cap with aluminium
seal. Each vial contains 1 dose of 0.3 mL, see Section 6.6 Special precautions for disposal and
other handling.
Pack size: 10 vials
Comirnaty LP.8.1 (Dark blue cap) 2.25 mL fill volume, 2 mL clear multidose vial (Type I
glass) with a stopper (synthetic bromobutyl rubber) and a Dark Blue flip-off plastic cap with
aluminium seal. Each vial contains 6 doses of 0.3 mL, see Section 6.6 Special precautions for
disposal and other handling.
Pack size: 10 vials
6.6 Special precautions for disposal and other handling
Comirnaty LP.8.1 Suspension for Injection (Blue caps)
Handing prior to use
Frozen vials must be completely thawed prior to use. Frozen vials should be transferred to 2 °C
to 8 °C to thaw. Thaw times for 10-vial packs are noted in table below:
Vial Cap Color
Time That May Be Required For a 10-vial Pack
to Thaw (at 2 °C to 8 °C)
Light Blue
2 hours
Dark Blue
6 hours
• Upon moving frozen vaccine to 2 °C to 8 °C storage, update the expiry date on the carton.
The updated expiry date should reflect 10 weeks from the date of transfer to refrigerated
conditions (2 °C to 8 °C) and not exceeding the original printed expiry date (EXP).
• Alternatively, individual frozen vials may be thawed for 30 minutes at temperatures up to
30 °C for immediate use.
• If the vaccine is received at 2 °C to 8 °C it should continue to be stored at 2 °C to 8 °C.
Check that the carton has been previously updated to reflect the 10-week refrigerated expiry
date.
• Unopened vials can be stored for up to 12 hours at temperatures up to 30 °C. Total storage
time between 8 ºC to 30 ºC, inclusive of storage before and after puncture, should not
exceed 24 hours.
Comirnaty LP.8.1 - Suspension for Injection (Blue caps)
Preparation for administration
Comirnaty LP.8.1 Suspension for Injection should be prepared by a healthcare professional
using aseptic technique to ensure the sterility of the prepared suspension.
Vials of Comirnaty LP.8.1 Suspension for Injection have a blue cap, contain either 1 or 6 doses
of 0.3 mL of vaccine and
do not require dilution.
o Light Blue cap: single dose vial
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 35 of 37
o Dark Blue cap: 6 dose multidose vial
Vial verification
Prior to administration, check the name and strength of the vaccine on the vial label and the
colour of the vial cap and vial label border to ensure it is the intended presentation. Check
whether the vial is a single dose vial or a multidose vial and check if the vial requires dilution.
• Check appearance of vaccine prior to mixing and administration.
o Blue cap vials: Prior to mixing, the vaccine is a clear to slightly opalescent
dispersion and may contain white to off-white opaque amorphous particles.
• Gently invert the vial 10 times.
Do not shake.
• Do not use the vaccine if particulates or discoloration are present after mixing.
Preparation of individual doses
• Using aseptic technique, cleanse the vial stopper with a single-use antiseptic swab.
• Withdraw a 0.3 mL single dose.
•
For Dark Blue cap multidose vials (6 doses per vial):
o After first puncture, record appropriate date and time on the vial and store at 2 ºC
to 30 ºC for up to 12 hours. Do not re-freeze.
o Each dose must contain 0.3 mL of vaccine. Low dead-volume syringes and/or
needles should be used in order to extract all doses from a single vial. The low
dead-volume syringe and needle combination should have a dead volume of no
more than 35 microlitres.
o If the amount of vaccine remaining in the vial cannot provide a full dose, discard
the vial and any excess volume.
Any unused medicine or waste material should be disposed of in accordance with local
requirements.
7. MEDICINE SCHEDULE
Prescription Medicine.
8. SPONSOR
Pfizer New Zealand Limited
P O Box 3998
Auckland, New Zealand
Toll Free Number: 0800 736 363
9. DATE OF FIRST APPROVAL
Date of publication in the New Zealand Gazette of consent to distribute this medicine:
30 October 2025
10. DATE OF REVISION OF THE TEXT
24 November 2025
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 36 of 37
Comirnaty® is a registered trademark of BioNTech SE. Used under license.
Summary of Updates
Section
Update
4.4
Addition of Study C4591024 data (immunocompromised)
4.6
Addition of Study C4591015 data (maternal study)
4.8
Addition of AE data from Study C4591024 & Study C4591015
Addition of data from Study C4591054 SSA & SSB
Addition of data from Study C4591048 SSE
4.9
Inclusion of post-authorisation experience
5.1
Addition of Study C4591024 & Study C4591015 clinical data
Addition of data from Study C4591054 SSA & SSB
Addition of data from Study C4591048 SSE
Version: pfdclpbi11125
Supersedes: pfdclpbi11025
Page 37 of 37
Document 4
NEW ZEALAND DATA SHEET
1. PRODUCT NAME
Comirnaty® LP.8.1 COVID-19 mRNA vaccine , 30 micrograms/0.3 mL dose, suspension for
injection (dark grey caps)
Comirnaty® LP.8.1 COVID-19 mRNA vaccine , 30 micrograms/0.3 mL dose, suspension for
injection in a pre-filled syringe
2. QUALITATIVE AND QUANTITATIVE COMPOSITION
This is a multidose vial (2.25 mL), or single dose pre-filled syringe. Do not dilute prior to
use.
One multidose vial (dark grey cap) contains 6 doses of 0.3 mL.
One pre-filled syringe (glass) contains 1 dose of 0.3 mL.
One dose (0.3 mL) contains 30 micrograms of SARS-CoV-2 spike protein (mRNA) LP.8.1, a
COVID-19 mRNA Vaccine (embedded in lipid nanoparticles).
SARS-CoV-2 spike protein (mRNA) LP.8.1 is a single-stranded, 5’-capped messenger RNA
(mRNA) produced using a cell-free
in vitro transcription from the corresponding DNA
templates, encoding the viral spike (S) protein of severe acute respiratory syndrome
coronavirus 2 (SARS-CoV-2) (LP.8.1).
For the full list of excipients, see Section 6.1 List of excipients.
3. PHARMACEUTICAL FORM
Suspension for injection.
The vaccine is a white to off-white suspension (pH 6.9 - 7.9).
4. CLINICAL PARTICULARS
4.1 Therapeutic indications
Active immunisation to prevent coronavirus disease 2019 (COVID-19) caused by SARS-CoV-
2, in individuals 12 years of age and older.
The use of this vaccine should be in accordance with official recommendations.
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 1 of 34
4.2 Dose and method of administration
Dose
Individuals 12 years of age and older
Comirnaty LP.8.1 is administered intramuscularly as a single dose of 0.3 mL for individuals
12 years of age and older, regardless of prior COVID-19 vaccination status.
For individuals who have previously been vaccinated with a COVID-19 vaccine, Comirnaty
LP.8.1 should be administered at least 3 months after the most recent dose of a COVID-19
vaccine.
Severely immunocompromised aged 12 years and older
Additional doses may be administered to individuals who are severely immunocompromised
in accordance with official recommendations (see Section 4.4 Special warnings and
precautions for use).
Paediatric population
There are paediatric formulations available for infants aged 6 months and above, and children
below 12 years of age. For details, please refer to the data sheets for other formulations. The
safety and efficacy of the vaccine in infants aged less than 6 months have not yet been
established.
Elderly population
No dosage adjustment is required in elderly individuals ≥65 years of age.
Method of administration
Comirnaty LP.8.1 should be administered intramuscularly. The preferred site of administration
is the deltoid muscle of the upper arm.
Do not inject Comirnaty LP.8.1 intravascularly, subcutaneously or intradermally.
Comirnaty should not be mixed in the same syringe with any other vaccines or medicinal
products.
For precautions to be taken before administering Comirnaty LP.8.1, see Section 4.4 Special
warnings and precautions for use. For instructions regarding thawing, handling and disposal of
the vaccine, see Section 6.6 Special precautions for disposal and other handling.
Multidose vials
Multidose vials of Comirnaty LP.8.1 (dark grey cap) contain 6 doses of 0.3 mL of vaccine and
do not require dilution.
In order to extract 6 doses from a multidose vial (dark grey cap), low dead-volume syringes
and/or needles should be used. The low dead-volume syringe and needle combination should
have a dead volume of no more than 35 microlitres. If standard syringes and needles are used,
there may not be sufficient volume to extract a sixth dose from a single vial. Irrespective of the
type of syringe and needle:
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 2 of 34
- Each dose must contain 0.3 mL of vaccine.
- If the amount of vaccine remaining in the vial cannot provide a full dose of 0.3 mL,
discard the vial and any excess volume.
- Do not pool excess vaccine from multiple vials.
For instructions on thawing, handling and dose preparation of Comirnaty LP.8.1 suspension
for injection, see Section 6.6 Special precautions for disposal and other handling.
Pre-filled syringes (glass)
The glass pre-filled syringes are supplied thawed and must not be shaken. If the glass pre-filled
syringe has been frozen, discard. Do not shake. For instructions on handling and thawing the
pre-filled syringes prior to use, refer to Section 6.6 Special precautions for disposal and other
handling.
Each single dose pre-filled syringe contains 1 dose of 0.3 mL of vaccine.
Remove tip cap and attach a sterile needle appropriate for intramuscular injection and
administer the entire volume of the syringe.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of
excipients.
4.4 Special warnings and precautions for use
Traceability
In order to improve the traceability of biological medicinal products, the name and the batch
number of the administered product should be clearly recorded.
General recommendations
Hypersensitivity and anaphylaxis
Events of anaphylaxis have been reported. Appropriate medical treatment and supervision
should always be readily available in case of an anaphylactic reaction following the
administration of Comirnaty.
The individual should be kept under close observation for at least 15 minutes following
vaccination. A second dose of Comirnaty should not be given to those who have experienced
anaphylaxis to the first dose of Comirnaty.
Myocarditis and pericarditis
Very rare cases of myocarditis and pericarditis have been observed following vaccination with
Comirnaty. These cases have primarily occurred within 14 days following vaccination, more
often after the second vaccination, and more often, but not exclusively in younger men. There
have been reports in females. Based on accumulating data, the reporting rates of myocarditis
and pericarditis after primary series in children ages 5 to 11 years are lower than in ages 12 to
17 years. Rates of myocarditis and pericarditis in booster doses do not appear to be higher than
after the second dose in the primary series. The cases are generally mild and individuals tend
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 3 of 34
to recover within a short time following standard treatment and rest. Cases of myocarditis and
pericarditis following vaccination have rarely been associated with severe outcomes including
death.
Healthcare professionals should be alert to the signs and symptoms of myocarditis and
pericarditis, including atypical presentations. Vaccinees should be instructed to seek
immediate medical attention if they develop symptoms indicative of myocarditis or pericarditis
such as (acute and persisting) chest pain, shortness of breath, or palpitations following
vaccination. Non-specific symptoms of myocarditis and pericarditis also include fatigue,
nausea and vomiting, abdominal pain, dizziness or syncope, oedema and cough. Healthcare
professionals should consult guidance and/or specialists to diagnose and treat this condition.
Stress-related responses
Some individuals may have stress-related responses associated with the process of vaccination
itself. Stress-related responses are temporary and resolve on their own. They may include
dizziness, fainting, palpitations, increases in heart rate, alterations in blood pressure, feeling
short of breath, tingling sensations, sweating and/or anxiety. Individuals should be advised to
bring symptoms to the attention of the vaccination provider for evaluation and precautions
should be in place to avoid injury from fainting.
Concurrent il ness
Vaccination should be postponed in individuals suffering from acute severe febrile illness or
acute infection. The presence of a minor infection and/or low grade fever should not delay
vaccination.
Thrombocytopenia and coagulation disorders
As with other intramuscular injections, the vaccine should be given with caution in individuals
receiving anticoagulant ther apy or those with thrombocytopenia or any coagulation disorder
(such as haemophilia) because bleeding or bruising may occur following an intramuscular
administration in these individuals.
Immunocompromised individuals
Immunocompromised persons, including individuals receiving immunosuppressant therapy,
may have a diminished immune response to the vaccine.
Clinical data on safety and immunogenicity after administration of Comirnaty (tozinameran)
in immunocompromised participants are available in 37 participants 2 to 4 years old,
65 participants 5 to 11 years old, 15 participants 12 to 17 years old, and 7 participants 18 years
of age and older (see Sections 4.8 Undesirable effects and 5.1 Pharmacodynamic properties).
Duration of protection
The duration of protection afforded by Comirnaty is unknown as it is still being determined by
ongoing clinical trials.
Limitations of vaccine effectiveness
As with any vaccine, vaccination with Comirnaty may not protect all vaccine recipients.
Individuals may not be fully protected until 7 days after their dose of Comirnaty.
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 4 of 34
Use in the elderly
Clinical studies of Comirnaty (tozinameran) include participants 65 years of age and older and
their data contributes to the overall assessment of safety and efficacy. See Section 5.1
Pharmacodynamic properties, Clinical trials, Efficacy against COVID-19. No dosage
adjustment is required in elderly individuals ≥65 years of age.
The data for use in the frail elderly is limited. The potential benefits of vaccination versus the
potential risk and clinical impact of even relatively mild systemic adverse events in the frail
elderly should be carefully assessed on a case-by-case basis.
The safety of a booster dose of Comirnaty (tozinameran) in individuals 65 years of age and
older is based on safety data in 12 booster dose recipients 65 to 85 years of age in Study
C4591001, 306 booster dose recipients 18 to 55 years of age in Study C4591001, and 1,175
booster dose recipients 65 years of age and older in Study C4591031. The effectiveness of a
booster dose of Comirnaty (tozinameran) in individuals 65 years of age and older is based on
effectiveness data in 306 booster dose recipients 18 to 55 years of age in Study C4591001, and
an efficacy analysis from participants 16 years of age and older in 9,945 participants in Study
C4591031.
Paediatric use
The safety and efficacy of Comirnaty in children aged less than 6 months of age have not yet
been established.
Effects on laboratory tests
No data available.
4.5 Interactions with other medicines and other forms of interactions
Comirnaty LP.8.1 (30 micrograms/dose only) may be administered concomitantly with
seasonal influenza vaccine.
The effectiveness and safety of concomitant Comirnaty (tozinameran) and seasonal influenza
vaccination in individuals > 65 years of age is extrapolated from Study C4591030 (see Section
5.1 Pharmacodynamic properties).
In individuals 18 years of age and older, Comirnaty may be administered concomitantly with
a pneumococcal conjugate vaccine (PCV) (see Section 5.1 Pharmacodynamic properties).
In individuals 60 years of age and older, Comirnaty may be administered concomitantly with
an unadjuvanted respiratory syncytial virus (RSV) vaccine (see Section 5.1 Pharmacodynamic
properties).
In individuals 65 years of age and older, Comirnaty may be administered concomitantly with
an RSV vaccine and a high dose influenza vaccine (see Section 5.1 Pharmacodynamic
properties).
Different injectable vaccines should be given at different injection sites.
Do not mix Comirnaty with other vaccines or products in the same syringe.
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 5 of 34
4.6 Fertility, pregnancy and lactation
Fertility
In a combined fertility and developmental toxicity study, female rats were intramuscularly
administered Comirnaty (tozinameran) prior to mating and during gestation (4 full human
doses of 30 micrograms each, spanning between pre-mating day 21 and gestation day 20).
SARS-CoV-2 neutralising antibodies were present in maternal animals from prior to mating to
the end of the study on postnatal day 21 as well as in fetuses and offspring. There were no
vaccine related effects on female fertility and pregnancy rate.
Pregnancy
No data are available yet regarding the use of Comirnaty LP.8.1 during pregnancy.
There are clinical study data from the use of Comirnaty (tozinameran) in 173 pregnant women
and no safety concerns were identified in the mother or their infant that were attributable to
maternal vaccination (see Section 4.8 Undesirable effects). Animal studies do not indicate
direct or indirect harmful effects with respect to pregnancy, embryo/fetal development,
parturition or post-natal development (see Section 4.6 Fertility, pregnancy and lactation,
Fertility).
Administration of Comirnaty LP.8.1 in pregnancy should only be considered when the
potential benefits outweigh any potential risks for the mother and fetus.
Lactation
No data are available yet regarding the use of Comirnaty LP.8.1 during breast-feeding. A
combined fertility and developmental toxicity study in rats did not show harmful effects on
offspring development before weaning (see Section 4.6 Fertility, pregnancy and lactation,
Fertility).
4.7 Effects on ability to drive and use machines
Comirnaty LP.8.1 has no, or negligible, influence on the ability to drive and use machines.
However, some of the effects mentioned under Section 4.8 Undesirable effects may
temporarily affect the ability to drive or use machines.
4.8 Undesirable effects
Summary of safety profile
The safety of Comirnaty (tozinameran) was evaluated in participants aged 6 months and older
in clinical studies (comprised of 22,026 participants 16 years of age and older and 1,131
adolescents 12 to 15 years of age from Study C4591001, and 3,109 children 5 to 11 years of
age, 2,368 participants 2 to 4 years of age and 1,458 participants 6 to 23 months of age from
Study C4591007) that have received at least one dose of Comirnaty (tozinameran).
Additionally, 306 existing Phase 3 participants at 18 to 55 years of age received a booster dose
of Comirnaty (tozinameran) approximately 6 months after the second dose in the non-placebo-
controlled booster dose portion of Study C4591001. The overall safety profile for the booster
dose was similar to that seen after 2 doses.
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 6 of 34
In Study C4591031, a placebo-controlled booster study, 5,081 participants 16 years of age and
older were recruited from Study C4591001 to receive a booster dose of Comirnaty
(tozinameran) at least 6 months after the second dose. The overall safety profile for the booster
dose was similar to that seen after 2 doses.
In a subset of Study C4591054 (Substudy A, Phase 2/3), 412 participants 12 years of age and
older, who had received at least 3 doses of an mRNA COVID-19 vaccine, received a booster
dose of Comirnaty Omicron XBB.1.5. In another substudy of Study C4591054 (Substudy B,
Phase 2/3), 311 participants 12 years of age and older, who were COVID-19 vaccine-naïve,
received 1 dose of Comirnaty Omicron XBB.1.5. The safety profile of Comirnaty Omicron
XBB.1.5 was similar to the overall Comirnaty safety profile.
In a subset of C4591048 (Substudy E, Phase 2/3), 310 participants 5 to 11 years of age who
were COVID-19 vaccine-naïve, received 1 dose of Comirnaty Omicron XBB.1.5. The safety
profile of Comirnaty Omicron XBB.1.5 was similar to the overall Comirnaty safety profile.
Omicron-adapted Comirnaty
Participants 12 years of age and older – after a single dose in vaccine-naïve individuals
In a subset of C4591054 (Substudy B, Phase 2/3), 311 participants 12 years of age and older,
who were considered to be baseline SARS-CoV-2 positive and COVID-19 vaccine-naïve,
received 1 dose of Comirnaty Omicron XBB.1.5 (raxtozinameran). Participants had a median
follow-up time of 6.4 months up to a data cut-off date of 23 April 2024.
The safety profile of Comirnaty Omicron XBB.1.5 was similar to the overall Comirnaty safety
profile. The most frequent adverse reactions in participants were injection site pain (>50%),
fatigue (>30%), headache (>20%), chills (>10%), diarrhea (>10%), new or worsened muscle
pain (>10%), new or worsened joint pain (>10%), and swelling (>10%).
Participants 5 to 11 years of age – after a single dose in vaccine-naïve individuals
In a subset of C4591048 (Substudy E, Phase 2/3), 310 participants 5 to 11 years of age who
were COVID-19 vaccine-naïve, received 1 dose of Comirnaty Omicron XBB.1.5. Participants
had a median follow-up time of 6.4 months up to a data cut-off date of 1 November 2024.
The safety profile of Comirnaty Omicron XBB.1.5 was similar to the overall Comirnaty safety
profile. The most frequent adverse reactions in participants were pain at the injection site
(>40%), fatigue (>10%), headache (>10%), and new or worsened muscle pain (>10%).
Participants 12 years of age and older – after a booster dose
In a subset of C4591054 (Substudy A, Phase 2/3), 412 participants 12 years of age and older,
who had received at least 3 doses of an authorized mRNA COVID-19 vaccine, received a
booster dose of Comirnaty Omicron XBB.1.5.The safety profile of Comirnaty Omicron
XBB.1.5 was similar to the overall Comirnaty safety profile.
COMIRNATY (tozinameran)
Participants 16 years of age and older – after 2 doses
In Study C4591001, a total of 22,026 participants 16 years of age or older received at least 1
dose of Comirnaty (tozinameran) 30 micrograms and a total of 22,021 participants 16 years of
age or older received placebo (including 138 and 145 adolescents 16 and 17 years of age in the
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 7 of 34
Comirnaty (tozinameran) and placebo groups, respectively). A total of 20,519 participants 16
years of age or older received 2 doses of Comirnaty (tozinameran).
At the time of the analysis of Study C4591001 with a data cut-off of 13 March 2021 for the
placebo-controlled blinded follow-up period up to the participants’ unblinding dates, a total of
25,651 (58.2%) participants (13,031 Comirnaty (tozinameran) and 12,620 placebo) 16 years of
age and older were followed up for ≥4 months after the second dose. This included a total of
15,111 (7,704 Comirnaty (tozinameran) and 7,407 placebo) participants 16 to 55 years of age
and a total of 10,540 (5,327 Comirnaty (tozinameran) and 5,213 placebo) participants 56 years
and older.
The most frequent adverse reactions in participants 16 years of age and older that received 2
doses were injection site pain (>80%), fatigue (>60%), headache (>50%), myalgia (>40%),
chills (>30%), arthralgia (>20%), pyrexia and injection site swelling (>10%) and were usually
mild or moderate in intensity and resolved within a few days after vaccination. A slightly lower
frequency of reactogenicity events was associated with greater age.
The safety profile in 545 subjects receiving Comirnaty (tozinameran), that were seropositive
for SARS-CoV-2 at baseline, was similar to that seen in the general population.
Study C4591001 also included 200 participants with confirmed stable human
immunodeficiency virus (HIV) infection. The safety profile of the participants receiving
Comirnaty (tozinameran) (n=100) in the individuals with stable HIV infection was similar to
that seen in the general population.
Adolescents 12 to 15 years of age – after 2 doses
In an analysis of long term safety follow-up in Study C4591001, 2,260 adolescents
[1,131 Comirnaty (tozinameran) 30 micrograms; 1,129 placebo] were 12 to 15 years of age.
Of these, 1,559 adolescents (786 Comirnaty (tozinameran) and 773 placebo) have been
followed for ≥ 4 months after the second dose of Comirnaty (tozinameran). The safety
evaluation in Study C4591001 is ongoing.
The most frequent adverse reactions in adolescents 12 to 15 years of age that received 2 doses
were injection site pain (>90%), fatigue and headache (>70%), myalgia and chills (>40%),
arthralgia and pyrexia (>20%).
Participants 12 years of age and older – after booster dose
A subset from Study C4591001 Phase 2/3 participants of 306 adults 18 to 55 years of age who
completed the original Comirnaty (tozinameran) 2-dose course, received a booster dose of
Comirnaty (tozinameran) approximately 6 months (range of 4.8 to 8.0 months) after receiving
Dose 2. Of these, 301 participants have been followed for ≥4 months after the booster dose of
Comirnaty (tozinameran).
The most frequent adverse reactions in participants 18 to 55 years of age were injection site
pain (>80%), fatigue (>60%), headache (>40%), myalgia (>30%), chills and arthralgia (>20%).
In Study C4591031, a placebo-controlled booster study, participants 16 years of age and older
recruited from Study C4591001 received a booster dose of Comirnaty (tozinameran) (5,081
participants), or placebo (5,044 participants) at least 6 months after the second dose of
Comirnaty (tozinameran). Overall, participants who received a booster dose, had a median
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 8 of 34
follow-up time of 2.8 months (range 0.3 to 7.5 months) after the booster dose in the blinded
placebo-controlled follow-up period to the cut-off date (8 February 2022). Of these, 1281
participants (895 Comirnaty (tozinameran) and 386 placebo) were followed for ≥ 4 months
after the booster dose of Comirnaty (tozinameran). The overall safety profile for the booster
dose was similar to that seen after 2 doses.
In another subset from Study C4591001, 825 adolescents 12 to 15 years of age who completed
the Comirnaty (tozinameran) 2-dose course, received a booster dose of Comirnaty
(tozinameran) approximately 11.2 months (range of 6.3 to 20.1 months) after receiving Dose
2. Overall, participants who received a booster dose, had a median follow-up time of 9.5
months (range 1.5 to 10.7 months) based on data up to the cut-off date (3 November 2022). No
new adverse reactions of Comirnaty (tozinameran) were identified.
Participants 18 years of age and older – after subsequent booster doses
In a subset from study C4591031 (Phase 3), 325 adults 18 to ≤55 years of age who had
completed 3 doses of Comirnaty (tozinameran), received a booster (fourth dose) of Comirnaty
(tozinameran 30 micrograms) 90 to 180 days after receiving Dose 3. Participants who received
a booster (fourth dose) of Comirnaty (tozinameran 30 micrograms) had a median follow-up
time of 1.4 months. The most frequent adverse reactions in these participants were injection
site pain (>70%), fatigue (>60%), headache (>40%), myalgia and chills (>20%) and arthralgia
(>10%).
In a subset from Study C4591031 (Phase 3), 305 adults greater than 55 years of age who had
completed 3 doses of Comirnaty (tozinameran), received a booster (fourth dose) of Comirnaty
(tozinameran 30 micrograms) 5.3 to 13.1 months after receiving Dose 3. Participants who
received a booster (fourth dose) of Comirnaty (tozinameran 30 micrograms) had a median
follow-up time of at least 1.7 months up to a data cutoff date of 16 May 2022. The most frequent
adverse reactions in participants greater than 55 years of age were injection site pain (60%),
fatigue (>40%), headache (>20%), myalgia and chills (>10%).
Tabulated list of adverse reactions from clinical studies and post-authorisation
experience
Adverse reactions observed during clinical studies are listed below according to the following
frequency categories:
Very common (≥ 1/10),
Common (≥ 1/100 to < 1/10),
Uncommon (≥ 1/1,000 to < 1/100),
Rare (≥ 1/10,000 to < 1/1,000),
Very rare (< 1/10,000),
Not known (cannot be estimated from the available data).
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 9 of 34
Table 1: Adverse reactions from Comirnaty (tozinameran) and Comirnaty Omicron
XBB.1.5 (raxtozinameran) clinical trials: Individuals 12 years of age and older
Rare
Not known
System Organ
Very
Common
Uncommon
(≥ 1/10,000
(cannot be
Class
common (≥ 1/100 to
(≥ 1/1,000 to
estimated from
(≥ 1/10)
< 1/10)
< 1/100)
to
< 1/1,000)
the available
data)
Blood and
Lymphadenopathya
lymphatic
system disorders
Metabolism and
Decreased appetite
nutrition
disorders
Psychiatric
Insomnia
disorders
Nervous system Headache
Lethargy
Acute
disorders
peripheral
facial
paralysisb
Gastrointestinal
Nausea;
disorders
Skin and
Hyperhidrosis;
subcutaneous
Night sweats
tissue disorders
Musculoskeletal Arthralgia;
and connective
Myalgia
tissue disorders
General
Injection
Injection
Asthenia; Malaise;
Facial swellingd
disorders and
site pain;
site redness
administration
Fatigue;
site conditions
Chills;
Pyrexiac;
Injection
site
swelling
a A higher frequency of lymphadenopathy (2.8% vs 0.4%) was observed in participants receiving a booster dose
in Study C4591031 compared to participants receiving 2 doses.
b Through the clinical trial safety follow-up period to 14 November 2020, acute peripheral facial paralysis (or
palsy) was reported by four participants in the Comirnaty (tozinameran) group. Onset was Day 37 after Dose 1
(participant did not receive Dose 2) and Days 3, 9, and 48 after Dose 2. No cases of acute peripheral facial paralysis
(or palsy) were reported in the placebo group.
c A higher frequency of pyrexia was observed after the second dose compared to the first dose. The preferred term
pyrexia is a cluster term covering also body temperature increased.
d Facial swelling in vaccine recipients with a history of injection of dermatological fillers
Special populations
Pregnant women and infants born to maternal participants – after 2 doses of Comirnaty
(tozinameran)
Study C4591015, a Phase 2/3, placebo-controlled study, evaluated Comirnaty (tozinameran)
or placebo administered in 2 doses, approximately 21 days apart, in pregnant women 18 years
of age and older, with the first dose given at 24 to 34 weeks gestation. A total of 346 pregnant
women received Comirnaty (tozinameran) (n=173) or placebo (n=173).
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 10 of 34
The most frequent adverse reactions in pregnant women who received any primary series dose
with Comirnaty (tozinameran) included injection site pain (>80%), fatigue (>60%),
headache (>50%), myalgia (>30%), chills, arthralgia, and injection site swelling (>10%).
The safety profile in pregnant women who received Comirnaty (tozinameran) was similar to
that of nonpregnant participants in other clinical studies, with no newly identified adverse
reactions.
In Study C4591015, safety in infants born to maternal participants who received Comirnaty
(tozinameran) (n=167) or placebo (n=168) was evaluated at birth and up to 6 months after birth.
No safety concerns were identified that were attributable to maternal vaccination with
Comirnaty (tozinameran).
Immunocompromised participants (adults and children)
In study C4591024, 37 participants 2 to 4 years old, 65 participants 5 to 11 years old, 15
participants 12 to 17 years old, and 7 participants 18 years of age and older from 5 different
immunocompromised disease subsets (immunomodulatory therapy, solid organ transplant,
stem cell transplant, non-small cell lung cancer (NSCLC)/chronic lymphocytic leukaemia
(CLL) and haemodialysis) received at least 1 and up to 4 doses of Comirnaty (tozinameran)
(Doses 1 and 2 were separated by 21 days, Doses 2 and 3 were separated by 28 days and Dose
4 was administered 3 to 6 months after Dose 3).
The safety profile in immunocompromised participants 2 years of age and older who received
Comirnaty (tozinameran) was similar to that in non-immunocompromised participants in other
clinical studies, with no newly identified adverse reactions.
Post-marketing experience
Although the events listed in Table 2 were not observed in the clinical trials, they are considered
adverse drug reactions for Comirnaty as they were reported in the post-marketing experience.
As these reactions were derived from spontaneous reports, the frequencies could not be
determined and are thus considered as not known.
Table 2: Adverse reactions from Comirnaty post marketing experience
System Organ Class
Adverse Drug Reaction
Immune system disorders
Anaphylaxis
Hypersensitivity reactions (e.g. rash, pruritis, urticaria, angioedema)
Cardiac disorders
Myocarditis
Pericarditis
Nervous system disorders
Dizziness
Gastrointestinal disorders
Diarrhoea
Vomiting
Musculoskeletal and connective Pain in extremity (arm)a
tissue disorders
General disorders and
Extensive swelling of vaccinated limb
administration site conditions
Reproductive system and breast Heavy menstrual bleedingb
disorders
a A higher frequency of pain in extremity (1.1% vs. 0.8%) was observed in participants receiving a booster dose in
Study C4591031 compared to participants receiving 2 doses.
b Most cases appear to be non-serious and temporary in nature.
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 11 of 34
Safety with concomitant vaccine administration
Concomitant administration with seasonal influenza vaccine
In Study C4591030, a Phase 3 study, participants 18 to 64 years of age who received Comirnaty
(tozinameran) coadministered with seasonal inactivated influenza vaccine (SIIV), quadrivalent
followed 1 month later by placebo (n=564), were compared to participants who received an
inactivated influenza vaccine with placebo followed 1 month later by Comirnaty (tozinameran)
alone (n=564). Reactogenicity events were reported more frequently by participants who
received Comirnaty (tozinameran) coadministered with SIIV, quadrivalent, compared to
participants who received Comirnaty (tozinameran) alone, but overall the reactogenicity events
were mostly mild to moderate in severity. The most common adverse reactions reported in the
coadministration group versus Comirnaty (tozinameran) alone were injection site pain (86.2%
vs 84.4%, respectively), fatigue (64.0% vs 50.8%, respectively) and headache (47.2% vs
37.8%, respectively).
Concomitant administration with pneumococcal conjugate vaccine
In Study B7471026, a Phase 3 study, participants 65 years of age and older who received a
booster dose of Comirnaty (tozinameran) coadministered with 20-valent pneumococcal
conjugate vaccine (20vPnC) (n=187), the
overall safety profile was similar with Comirnaty
(tozinameran) given alone (n=185). Overall, reactogenicity events were mostly mild to
moderate in severity. The most common adverse reactions reported in the coadministration
group versus Comirnaty (tozinameran) alone were injection site pain (72.4% vs 67.6%,
respectively), fatigue (54.1% vs 54.6%, respectively), and myalgia (32.4% vs 31.9 %,
respectively).
Concomitant administration with an RSV vaccine or with an RSV vaccine and a high
dose influenza vaccine
In Study C5481001, a Phase 1/2 study, participants 65 years of age and older who received
Comirnaty Original/Omicron BA.4-5 (tozinameran/famtozinameran) and RSV (bivalent,
recombinant) vaccine coadministered in one arm plus high dose quadrivalent influenza
vaccine (QIV) (n=158) or placebo (n=157) in the opposite arm were compared to participants
who received the individual vaccines given with placebo. The overall safety profile was
similar with Comirnaty Original/Omicron BA.4-5 given alone (n=150).
Overall, reactogenicity events reported for the concomitantly administered vaccines were
mostly mild to moderate in severity. The most common reported adverse reactions in the
Comirnaty Original/Omicron BA.4-5 administered concomitantly with RSV vaccine group,
Comirnaty Original/Omicron BA.4-5 administered concomitantly with both RSV vaccine and
high dose QIV group, and Comirnaty Original/Omicron BA.4-5 alone were injection site pain
(56.7%, 53.8%, and 62.7%, respectively) and fatigue (38.9%, 46.8%, and 35.3%, respectively)
.
Reporting suspected adverse effects
Reporting suspected adverse reactions after authorisation of the medicine is important. It allows
continued monitoring of the benefit/risk balance of the medicine. Healthcare professionals are
asked to report any suspected adverse reactions at
https://pophealth.my.site.com/carmreportnz/s/.
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 12 of 34
4.9 Overdose
In clinical trials, participants who received up to 2 times the recommended dose of Comirnaty
did not have an increase in reactogenicity or adverse reactions.
In post-authorisation experience, there have been reports of higher than recommended doses
of Comirnaty. In general, adverse events reported with overdoses have been similar to the
known adverse reaction profile of Comirnaty.
In the event of overdose, monitoring of vital functions and individualised symptomatic
treatment is recommended.
For risk assessment and advice on the management of overdose please contact the National
Poisons Centre on 0800 POISON (0800 764766).
5. PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: vaccines, Covid-19 RNA-based vaccines, ATC code: J07BN01.
Mechanism of action
The nucleoside-modified messenger RNA in Comirnaty is formulated in lipid nanoparticles,
which enable delivery of the non-replicating RNA into host cells to direct transient expression
of the SARS-CoV-2 spike (S) antigen. The mRNA codes for membrane-anchored, full-length
S with two point mutations within the central helix. Mutation of these two amino acids to
proline locks S in an antigenically preferred prefusion conformation. Comirnaty elicits both
neutralising antibody and cellular immune responses to the antigen, which may contribute to
protection against COVID-19.
Clinical efficacy and immunogenicity
Omicron-adapted Comirnaty
Immunogenicity in participants 12 years and older – after a single dose in vaccine-naïve
individuals
In a subset from C4591054, (Substudy B [Phase 2/3]), the evaluable immunogenicity
population of 302 vaccine-naïve participants 12 years of age and older who were considered to
be SARS−CoV-2 positive at baseline, received 1 dose of Comirnaty Omicron XBB.1.5, was
compared with participants in Substudy A [a subset from C4591054, (Phase 2/3)], who
received Comirnaty Omicron XBB.1.5 after at least 3 doses of an mRNA COVID-19 vaccine.
Neutralising titres against Omicron XBB.1.5 increased from baseline to 1 month after study
vaccination and were greater in participants receiving Comirnaty Omicron XBB.1.5 as a single
dose compared with participants who received Comirnaty Omicron XBB.1.5 after at least 3
doses of an mRNA COVID-19 vaccine. Noninferiority was met with respect to the geometric
mean ratio (GMR) of Omicron XBB.1.5-neutralising titres, and the difference in seroresponse
to the XBB.1.5 strain in Substudy B vaccine-naïve participants compared to the subset of
Substudy A (Table 3 and Table 4).
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 13 of 34
Table 3. Model-Based Geometric Mean Ratio – C4591054 Substudy B and Subset of
Substudy A – Evaluable Immunogenicity Population
Vaccine Group (as Assigned)
Group Comparison
Vaccine-Naïve
Vaccine-Experienced
Substudy B
Substudy A
Comirnaty
Comirnaty
Substudy B /
Omicron XBB.1.5
Omicron XBB.1.5
Substudy A
Sampling
30 mcg
30 mcg
Time
GMTc
GMTc
GMRd
Assaye
Pointa
nb
(95% CIc)
nb
(95% CIc)
(95% CId)
SARS-CoV-2
neutralisation
assay - Omicron
XBB.1.5 - NT50
4373.4
2915.7
1.93
(titre)e
1 month 299 (3757.1, 5090.9) 296
(2462.4, 3452.5)
(1.52, 2.44)f
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre;
LLOQ = lower limit of quantitation; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute respiratory
syndrome coronavirus 2.
a. Protocol-specified timing for blood sample collection.
b. n = Number of participants with valid and determinate assay results for the specified assay at both the
pre−vaccination time point and the given sampling time point.
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 ×
LLOQ.
d. GMRs and the corresponding 2-sided 95% CIs were calculated by exponentiating the difference in least
square means and the corresponding CIs based on a linear regression model with baseline assay results (log
scale), age, and vaccine group as covariates.
e. SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (Omicron subvariant
XBB.1.5).
f. Noninferiority is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67.
Table 4. Adjusted Difference in Percentages of Participants With Seroresponse –
C4591054 Substudy B and Subset of Substudy A – Evaluable Immunogenicity
Population
Vaccine Group (as Assigned)
Group Comparison
Vaccine-Naïve
Vaccine-Experienced
Substudy B
Substudy A
Comirnaty
Comirnaty
Omicron XBB.1.5
Omicron XBB.1.5
SARS-CoV-2
30 mcg
30 mcg
Adjusted Difference
Neutralisation Sampling
nc (%)
nc (%)
Differenc
Assayg
Time Pointa Nb
(95% CId)
Nb
(95% CId)
e %e
(95% CIf)
SARS-CoV-2
neutralisation
assay - Omicron
XBB.1.5 - NT50
253 (84.9)
218 (73.9)
(titre)g
1 month 298
(80.3, 88.8)
295
(68.5, 78.8)
7.31
(1.34, 13.28)h
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 14 of 34
Abbreviations: CI = confidence interval; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute respiratory
syndrome coronavirus 2.
a. Protocol-specified timing for blood sample collection.
b. N = number of participants with valid and determinate assay results for the specified assay at both the pre-
vaccination time point and the given sampling time point. These values are the denominators for the
percentage calculations.
c. n = Number of participants with a seroresponse for the given assay at the given sampling time point.
d. Exact 2-sided CI, based on the Clopper and Pearson method.
e. Difference in proportions, expressed as a percentage.
f. 2-Sided CI, based on the Miettinen and Nurminen method stratified by baseline neutralising titre category
(< median, ≥ median) and age group (< median, ≥ median). The median of baseline neutralising titres and
median age was calculated based on the pooled data in 2 comparator groups.
g. SARS-CoV-2 NT50 were determined using a validated 384-well assay platform Omicron subvariant
XBB.1.5).
h. Noninferiority is declared if the lower bound of the 2-sided 95% CI for the difference in percentages of
participants with seroresponse is >-10%.
Immunogenicity in participants 12 years of age and older – after a booster dose
In a subset from C4591054 (Substudy A, Phase 2/3), the evaluable immunogenicity population
included 382 participants 12 years of age and older who had previously received at least 3 prior
doses of an authorized mRNA COVID-19 vaccine, with the most recent dose being an Omicron
BA.4/BA.5-adapted bivalent vaccine, received a booster dose of Comirnaty Omicron XBB.1.5.
At baseline, 78.8% of participants were considered to be positive for prior SARS−CoV−2
infection.
Compared to participants receiving Comirnaty Original/Omicron BA.4-5 (C4591044),
participants receiving Comirnaty Omicron XBB.1.5 (C4591054) had higher GMTs against
Omicron XBB.1.5 (2622.3 [CI: 2246.6, 3060.9] versus 601.0 [CI: 499.5, 723.1]) and against
Omicron BA.4/BA.5 (5105.1 [CI: 4483.4, 5813.0] versus 4146.0 [CI: 3512.6, 4893.5]) at 1
month after vaccination.
Seroresponse (NT50) was higher against Omicron XBB.1.5, and lower against Omicron
BA.4/BA.5 among participants who received Comirnaty Omicron XBB.1.5 at 1 month after
vaccination compared to the participants who Comirnaty Original/Omicron BA.4-5
(C4591044) with NT50 against Omicron XBB.1.5 of 73.9% (CI: 69.2%, 78.3%) versus
52.8% (CI: 45.6%, 59.9%), and NT50 against Omicron BA.4/BA.5 of 48.3% (CI: 43.2%,
53.4%) versus 63.0% (CI: 55.9%, 69.7%).
Comirnaty (tozinameran)
Study C4591001 is a multicentre, multinational, Phase 1/2/3 randomised, placebo-controlled,
observer-blind dose-finding, vaccine candidate selection and efficacy study in participants 12
years of age and older. Randomisation was stratified by age: 12 to 15 years of age, 16 to 55
years of age, or 56 years of age and older, with a minimum of 40% of participants in the ≥56-
year stratum. The study excluded participants who were immunocompromised and those who
had previous clinical or microbiological diagnosis of COVID-19. Participants with pre-existing
stable disease, defined as disease not requiring significant change in therapy or hospitalisation
for worsening disease during the 6 weeks before enrolment, were included as were participants
with known stable infection with HIV, hepatitis C virus (HCV) or hepatitis B virus (HBV).
Efficacy in participants 16 years of age and older – after 2 doses
In the Phase 2/3 portion of Study C4591001, based on data accrued through
14 November 2020, approximately 44,000 participants were randomised equally and were to
receive 2 doses of Comirnaty (tozinameran) or placebo. The efficacy analyses included
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 15 of 34
participants that received their second vaccination within 19 to 42 days after their first
vaccination. The majority (93.1%) of vaccine recipients received the second dose 19 days to
23 days after Dose 1. Participants are planned to be followed for up to 24 months after Dose 2,
for assessments of safety and efficacy against COVID-19. In the clinical study, participants
were required to observe a minimum interval of 14 days before and after administration of an
influenza vaccine in order to receive either placebo or Comirnaty (tozinameran). In the clinical
study, participants were required to observe a minimum interval of 60 days before or after
receipt of blood/plasma products or immunoglobulins through to conclusion of the study in
order to receive either placebo or Comirnaty (tozinameran).
The population for the analysis of the primary efficacy endpoint included 36,621 participants
12 years of age and older (18,242 in the Comirnaty (tozinameran) group and 18,379 in the
placebo group) who did not have evidence of prior infection with SARS-CoV-2 through 7 days
after the second dose. In addition, 134 participants were between the ages of 16 to 17 years of
age (66 in the Comirnaty (tozinameran) group and 68 in the placebo group) and 1616
participants 75 years of age and older (804 in the Comirnaty (tozinameran) group and 812 in
the placebo group).
At the time of the primary efficacy analysis, participants had been followed for symptomatic
COVID-19 for in total 2,214 person-years for the Comirnaty (tozinameran) group and in total
2,222 person-years for the placebo group.
There were no meaningful clinical differences in overall vaccine efficacy in participants who
were at risk of severe COVID-19 including those with 1 or more comorbidities that increase
the risk of severe COVID-19 (e.g. asthma, body mass index (BMI) ≥30 kg/m2, chronic
pulmonary disease, diabetes mellitus, hypertension).
Comirnaty (tozinameran) efficacy information is presented in Table 5.
Table 5: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2, by age
subgroup – participants without evidence of infection prior to 7 days after Dose 2 –
evaluable efficacy (7 days) population
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence of prior
SARS-CoV-2 infection*
Comirnaty
(tozinameran)
Placebo
Vaccine efficacy
Subgroup
Na = 18,198
Na = 18,325
%
Cases n1b
Cases n1b
(95% CI)f
Surveillance timec (n2d)
Surveillance timec (n2d)
All participantse
8
162
95.0
2.214 (17,411)
2.222 (17,511)
(90.0, 97.9)
16 to 64 years
7
143
95.1
1.706 (13,549)
1.710 (13,618)
(89.6, 98.1)
65 years and older
1
19
94.7
0.508 (3848)
0.511 (3880)
(66.7, 99.9)
65 to 74 years
1
14
92.9
0.406 (3074)
0.406 (3095)
(53.1, 99.8)
75 years and older
0
5
100.0
0.102 (774)
0.106 (785)
(-13.1, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 [*Case definition: (at least 1 of) fever, new or increased cough,
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 16 of 34
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence of prior
SARS-CoV-2 infection*
Comirnaty
(tozinameran)
Placebo
Vaccine efficacy
Subgroup
Na = 18,198
Na = 18,325
%
Cases n1b
Cases n1b
(95% CI)f
Surveillance timec (n2d)
Surveillance timec (n2d)
new or increased shortness of breath, chills, new or increased muscle pain, new loss of taste or smell, sore
throat, diarrhoea or vomiting.]
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the last dose) of
past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not
detected by nucleic acid amplification tests (NAAT) [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of
the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. No confirmed cases were identified in adolescents 12 to 15 years of age.
f. Two-sided confidence interval (CI) for vaccine efficacy (VE) is derived based on the Clopper and Pearson
method adjusted to the surveillance time. CI not adjusted for multiplicity.
In the second primary analysis, efficacy of Comirnaty (tozinameran) in preventing first
COVID-19 occurrence from 7 days after Dose 2 compared to placebo was 94.6% (95% credible
interval of 89.9% to 97.3%) in participants 16 years of age and older with or without evidence
of prior infection with SARS-CoV-2.
Additionally, subgroup analyses of the primary efficacy endpoint showed similar efficacy point
estimates across genders, ethnic groups, and participants with medical comorbidities associated
with high risk of severe COVID-19.
Updated efficacy analyses were performed with additional confirmed COVID-19 cases accrued
during blinded placebo-controlled follow-up through 13 March 2021, representing up to
6 months of follow-up after Dose 2 for participants in the efficacy population.
The updated vaccine efficacy information is presented in Table 6.
Table 6: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2, by age
subgroup – participants without evidence of infection prior to 7 days after Dose 2 –
evaluable efficacy (7 days) population during the placebo-controlled follow-up period
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
of prior SARS-CoV-2 infection*
Comirnaty
(tozinameran)
Placebo
Na=20,998
Na=21,096
Vaccine efficacy
Cases n1b
Cases n1b
%
Subgroup
Surveil ance Timec (n2d)
Surveil ance Timec (n2d)
(95% CIe)
All participantsf
77
850
91.3
6.247 (20,712)
6.003 (20,713)
(89.0, 93.2)
16 to 64 years
70
710
90.6
4.859 (15,519)
4.654 (15,515)
(87.9, 92.7)
65 years and older
7
124
94.5
1.233 (4192)
1.202 (4226)
(88.3, 97.8)
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 17 of 34
65 to 74 years
6
98
94.1
0.994 (3350)
0.966 (3379)
(86.6, 97.9)
75 years and older
1
26
96.2
0.239 (842)
0.237 (847)
(76.9, 99.9)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative
at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Two-sided confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method
adjusted to the surveillance time.
f. Included confirmed cases in participants 12 to 15 years of age: 0 in the Comirnaty (tozinameran) group (both
without and with or without evidence of prior SARS-CoV-2 infection); 16 and 18 in the placebo group
(without and with or without evidence of prior SARS-CoV-2 infection, respectively).
Efficacy against severe COVID-19 in participants 12 years of age or older – after 2 doses
As of 13 March 2021, vaccine efficacy against severe COVID-19 is presented only for
participants with or without prior SARS-CoV-2 infection (Table 7) as the COVID-19 case
counts in participants without prior SARS-CoV-2 infection were the same as those in
participants with or without prior SARS-CoV-2 infection in both the Comirnaty (tozinameran)
and placebo groups.
Table 7. Vaccine Efficacy – First Severe COVID-19 Occurrence in Participants With
or Without* Prior SARS-CoV-2 Infection Based on Food and Drug Administration
(FDA)† Definition After Dose 1 or From 7 Days After Dose 2 in the Placebo-Controlled
Follow-up
Comirnaty (tozinameran)
Placebo
Cases n1a
Cases n1a
Vaccine Efficacy %
Surveil ance Time (n2b) Surveil ance Time (n2b)
(95% CIc)
1
30
96.7
After Dose 1d
8.439e (22,505)
8.288e (22,435)
(80.3, 99.9)
1
21
95.3
7 days after Dose 2f
6.522g (21,649)
6.404g (21,730)
(70.9, 99.9)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at
least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or increased
shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat; diarrhoea;
vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative
at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
† Severe illness from COVID-19 as defined by FDA is confirmed COVID-19 and presence of at least 1 of the
following:
• Clinical signs at rest indicative of severe systemic illness (respiratory rate ≥30 breaths per minute, heart
rate ≥125 beats per minute, saturation of oxygen ≤93% on room air at sea level, or ratio of arterial
oxygen partial pressure to fractional inspired oxygen <300 mm Hg);
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 18 of 34
• Respiratory failure [defined as needing high-flow oxygen, noninvasive ventilation, mechanical
ventilation or extracorporeal membrane oxygenation (ECMO)];
• Evidence of shock (systolic blood pressure <90 mm Hg, diastolic blood pressure <60 mm Hg, or
requiring vasopressors);
• Significant acute renal, hepatic, or neurologic dysfunction;
• Admission to an Intensive Care Unit;
• Death.
a. n1 = Number of participants meeting the endpoint definition.
b. n2 = Number of participants at risk for the endpoint.
c. Two-side confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method
adjusted to the surveillance time.
d. Efficacy assessed based on the Dose 1 all available efficacy (modified intention-to-treat) population that
included all randomised participants who received at least 1 dose of study intervention.
e. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from Dose 1 to the end of the surveillance
period.
f. Efficacy assessed based on the evaluable efficacy (7 Days) population that included al eligible randomised
participants who receive all dose(s) of study intervention as randomised within the predefined window, have
no other important protocol deviations as determined by the clinician
g. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
Efficacy and immunogenicity in adolescents 12 to 15 years of age – after 2 doses
An analysis of Study C4591001 has been performed in adolescents 12 to 15 years of age up to
a data cutoff date of 13 March 2021.
The vaccine efficacy information in adolescents 12 to 15 years of age is presented in Table 8.
Table 8: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2 –
participants without evidence of infection and with or without evidence of infection prior
to 7 days after Dose 2 – adolescents 12 to 15 years of age evaluable efficacy (7 days)
population
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
without evidence of prior SARS-CoV-2 infection*
Comirnaty
(tozinameran)
Placebo
Na = 1005
Na = 978
Cases n1b
Cases n1b
Vaccine efficacy
Surveil ance timec (n2d) Surveil ance timec (n2d)
% (95% CIe)
Adolescents
0
16
12 to 15 years
0.154 (1001)
0.147 (972)
100.0 (75.3, 100.0)
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
with or without* evidence of prior SARS-CoV-2 infection
Comirnaty
(tozinameran)
Placebo
Na = 1119
Na = 1110
Cases n1b
Cases n1b
Vaccine efficacy
Surveil ance timec (n2d) Surveil ance timec (n2d)
% (95% CIe)
Adolescents
0
18
12 to 15 years
0.170 (1109)
0.163 (1094)
100.0 (78.1, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at
least 1 symptom consistent with COVID-19 [*Case definition: (at least 1 of) fever, new or increased cough, new
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 19 of 34
or increased shortness of breath, chills, new or increased muscle pain, new loss of taste or smell, sore throat,
diarrhoea or vomiting).
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the last dose) of
past SARS-CoV-2 infection (i.e, N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not
detected by nucleic acid amplification tests (NAAT) [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all subjects within each group at
risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of subjects at risk for the endpoint.
e. Confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method adjusted
for surveillance time. CI not adjusted for multiplicity.
In Study C4591001 an analysis of SARS-CoV-2 neutralising titres in a randomly selected
subset of participants was performed to demonstrate non-inferior immune responses (within
1.5-fold) comparing adolescents 12 to 15 years of age to participants 16 to 25 years of age who
had no serological or virological evidence of past SARS-CoV-2 infection. The immune
response to Comirnaty (tozinameran) in adolescents 12 to 15 years of age (n = 190) was non-
inferior to the immune response in participants 16 to 25 years of age (n = 170), based on results
for SARS-CoV-2 neutralising titres at 1 month after Dose 2. The geometric mean titres (GMT)
ratio of the adolescents 12 to 15 years of age group to the participants 16 to 25 years of age
group was 1.76, with a 2-sided 95% CI of 1.47 to 2.10, meeting the 1.5-fold non-inferiority
criterion (the lower bound of the 2-sided 95% CI for the geometric mean ratio [GMR] >0.67),
which indicates a statistically greater response in the adolescents 12 to 15 years of age than that
of participants 16 to 25 years of age.
An updated efficacy analysis of Study C4591001 has been performed in approximately 2,260
adolescents 12 to 15 years of age evaluating confirmed COVID-19 cases accrued up to a data
cut-off date of 2 September 2021, representing up to 6 months of follow-up after Dose 2 for
participants in the efficacy population.
The updated vaccine efficacy information in adolescents 12 to 15 years of age is presented in
Table 9.
Table 9: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Dose 2:
Without Evidence of Infection and With or Without Evidence of Infection
Prior to 7 Days After Dose 2 – Blinded Placebo-Controlled Follow-up Period,
Adolescents 12 To 15 Years of Age Evaluable Efficacy (7 Days) Population
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age without
evidence of prior SARS-CoV-2 infection*
Comirnaty (tozinameran)
Placebo
Na=1057
Na=1030
Cases n1b
Cases n1b
Vaccine Efficacy %
Surveil ance Timec (n2d) Surveil ance Timec (n2d)
(95% CIe)
Adolescents
0
28
100.0
12 to 15 years of age
0.343 (1043)
0.322 (1019)
(86.8, 100.0)
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 20 of 34
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
with or without evidence of prior SARS-CoV-2 infection
Comirnaty (tozinameran)
Placebo
Na=1119
Na=1109
Cases n1b
Cases n1b
Vaccine Efficacy %
Surveil ance Timec (n2d) Surveil ance Timec (n2d)
(95% CIe)
Adolescents
0
30
100.0
12 to 15 years of age
0.362 (1098)
0.345 (1088)
(87.5, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at
least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or increased
shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat; diarrhoea;
vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at
Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT (nasal
swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group at
risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Two-sided confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method
adjusted for surveillance time.
Immunogenicity in participants 18 years of age and older – after booster dose
Effectiveness of a booster dose of Comirnaty (tozinameran) was based on an assessment of
50% neutralising titres (NT50) against SARS-CoV-2 (USA_WA1/2020). In Study C4591001,
analyses of NT50 1 month after the booster dose compared to 1 month after the primary series
in individuals 18 to 55 years of age who had no serological or virological evidence of past
SARS-CoV-2 infection up to 1 month after the booster vaccination demonstrated
noninferiority for both GMR and difference in seroresponse rates. Seroresponse for a
participant was defined as achieving a ≥4-fold rise in NT50 from baseline (before Dose 1),
These analyses are summarised in Table 10.
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 21 of 34
Table 10. SARS-CoV-2 neutralisation assay - NT50 (titre)† (SARS-CoV-2
USA_WA1/2020) – GMT and seroresponse rate comparison of 1 month after booster dose
to 1 month after primary series – participants 18 to 55 years of age without evidence of
infection up to 1 month after booster dose* – booster dose evaluable immunogenicity
population±
1 month
after booster
dose/-
1 month
after
Met
1 month after
1 month after
primary
noninferiority
booster dose
primary series
series
objective
n
(95% CI)
(95% CI)
(97.5% CI)
(Y/N)
Geometric mean
50% neutralising
2466.0b
755.7
b
3.26c
titre (GMTb)
212a (2202.6, 2760.8) (663.1, 861.2)
(2.76, 3.86)
Yd
Seroresponse rate
199f
190f
(%) for 50%
99.5%
95.0%
4.5%g
neutralising titre† 200e (97.2%, 100.0%) (91.0%, 97.6%) (1.0%, 7.9%
h)
Yi
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre;
LLOQ = lower limit of quantitation; N-binding = SARS-CoV-2 nucleoprotein-binding; NAAT = nucleic acid
amplification test; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute respiratory syndrome
coronavirus 2; Y/N = yes/no.
† SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation
Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus
neutralisation is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum
dilution at which 50% of the virus is neutralised.
* Participants who had no serological or virological evidence (up to 1 month after receipt of a booster dose
of Comirnaty) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative and
SARS-CoV-2 not detected by NAAT [nasal swab]) and had a negative NAAT (nasal swab) at any
unscheduled visit up to 1 month after the booster dose were included in the analysis.
± All eligible participants who had received 2 doses of Comirnaty (tozinameran) as initially randomised,
with Dose 2 received within the predefined window (within 19 to 42 days after Dose 1), received a booster
dose of Comirnaty (tozinameran), had at least 1 valid and determinate immunogenicity result after booster
dose from a blood collection within an appropriate window (within 28 to 42 days after the booster dose),
and had no other important protocol deviations as determined by the clinician.
a. n = Number of participants with valid and determinate assay results at both sampling time points within
specified window.
b. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
c. GMRs and 2-sided 97.5% CIs were calculated by exponentiating the mean differences in the logarithms of
the assay and the corresponding CIs (based on the Student t distribution).
d. Noninferiority is declared if the lower bound of the 2-sided 97.5% CI for the GMR is > 0.67 and the point
estimate of the GMR is ≥ 0.80.
e. n = Number of participants with valid and determinate assay results for the specified assay at baseline,
1 month after Dose 2 and 1 month after the booster dose within specified window. These values are the
denominators for the percentage calculations.
f. Number of participants with seroresponse for the given assay at the given dose/sampling time point. Exact
2-sided CI based on the Clopper and Pearson method.
g. Difference in proportions, expressed as a percentage (1 month after booster dose – 1 month after Dose 2).
h. Adjusted Wald 2-sided CI for the difference in proportions, expressed as a percentage.
i. Noninferiority is declared if the lower bound of the 2-sided 97.5% CI for the percentage difference is
> -10%.
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 22 of 34
Relative vaccine efficacy in participants 16 years of age and older – after booster dose
An interim efficacy analysis of Study C4591031, a placebo-controlled booster study, was
performed in approximately 10,000 participants 16 years of age and older who were recruited
from Study C4591001, evaluated confirmed COVID-19 cases accrued from at least 7 days after
booster vaccination up to a data cut-off date of 8 February 2022 (a period when Delta and then
Omicron was the predominant variant), which represents a median of 2.8 months (range 0.3 to
7.5 months) post-booster follow-up. Vaccine efficacy of the Comirnaty (tozinameran) booster
dose after the primary series relative to the placebo booster group who only received the
primary series dose was assessed. The relative vaccine efficacy information for participants
16 years of age and older is presented in Table 11.
Table 11: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Booster
Vaccination – Participants 16 Years of Age and Older Without Evidence of Infection
and Participants With or Without Evidence of Infection Prior to 7 Days After Booster
Vaccination – Evaluable Efficacy Population
First COVID-19 occurrence from 7 days after booster dose in participants without evidence
of prior SARS-CoV-2 infection*
Comirnaty
(tozinameran)
Placebo
Na=4689
Na=4664
Relative Vaccine
Cases n1b
Cases n1b
Efficacye %
Surveil ance Timec (n2d) Surveil ance Timec (n2d)
(95% CIf)
First COVID-19
occurrence from
7 days after booster
63
148
63.9
vaccination
1.098 (4639)
0.932 (4601)
(51.1, 73.5)
First COVID-19 occurrence from 7 days after booster dose in participants with or without
evidence of prior SARS-CoV-2 infection
Comirnaty
(tozinameran)
Placebo
Na=4977
Na=4942
Relative Vaccine
Cases n1b
Cases n1b
Efficacye %
Surveil ance Timec (n2d) Surveil ance Timec (n2d)
(95% CIf)
First COVID-19
occurrence from
7 days after booster
67
150
62.4
vaccination
1.173 (4903)
0.989 (4846)
(49.5, 72.2)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the booster
vaccination) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and
SARS-CoV-2 not detected by NAAT [nasal swab] at Visit 1, and had a negative NAAT [nasal swab] at
any unscheduled visit prior to 7 days after booster vaccination) were included in the analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each
group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after the booster
vaccination to the end of the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Relative vaccine efficacy of the Comirnaty (tozinameran) booster group relative to the placebo group
(non-booster).
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 23 of 34
f. Two-sided confidence interval (CI) for relative vaccine efficacy is derived based on the Clopper and
Pearson method adjusted for surveillance time.
Immunogenicity in pregnant women and infants born to maternal participants – after 2
doses with Comirnaty (tozinameran)
Study C4591015 was a Phase 2/3 multinational, placebo-controlled, observer-blind study that
enrolled pregnant women 18 years of age and older to receive 2 doses of Comirnaty
(tozinameran) (n=173) or placebo (n=173). Pregnant women received Dose 1 of Comirnaty
(tozinameran) at 24 to 34 weeks gestation and the majority (90.2%) received the second dose
19 to 23 days after Dose 1.
Descriptive immunogenicity analysis was performed in pregnant women receiving Comirnaty
(tozinameran) in Study C4591015 compared to a comparator subset of nonpregnant women
from Study C4591001 evaluating the ratio of the neutralising GMT (GMR) 1 month after Dose
2.
The evaluable immunogenicity population who received Comirnaty (tozinameran) in the
maternal participants group in Study C4591015 (n=111) and in nonpregnant participants in
Study C4591001 (n=114) comprised of 69.4% vs. 82.5% White, 27.0% vs. 5.3% Black or
African American, 1.8% vs. 6.1% Asian, 0 vs 4.4% multiracial participants, 37.8% vs 34.2%
Hispanic/Latino, 37.8% vs 3.5% had a positive baseline SARS-CoV-2 status, and 38.7% vs
27.2% were obese [BMI ≥30 kg/m2 (pre-pregnancy weight in participants in Study
C4591015)], respectively. In maternal participants group in Study C4591015 and in
nonpregnant participants in Study C4591001 who received Comirnaty (tozinameran), the
median age was 30 years (range 18 to 44 years of age) in both groups.
The immunogenicity results after 2 doses of Comirnaty (tozinameran) in pregnant women
18 years of age and older are presented in Table 12.
Table 12. Geometric Mean Ratios – Participants Without* or With or Without Evidence
of Infection up to 1 Month After Dose 2 – Maternal Participants (Study
C4591015) and Nonpregnant Female Participants (Study C4591001) –
Evaluable Immunogenicity Population
Participants Without Evidence of Infection*
Comirnaty (tozinameran)
Study C4591015
Study C4591001
Pregnant/
Pregnant Women
Nonpregnant Women Nonpregnant
Dose/
Sampling
GMTd
GMTd
GMRe
Assay
Time Pointb nc
(95% CId)
nc
(95% CId)
(95% CI)e
SARS-CoV-2
neutralisation
assay - NT50
1109.2
1663.7
0.67
(titre)a
2/1 month
58
(849.2, 1448.9) 107 (1411.5, 1960.8)
(0.50, 0.90)
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 24 of 34
Participants With or Without Evidence of Infection
Comirnaty (tozinameran)
Study C4591015
Study C4591001
Pregnant/
Pregnant Women
Nonpregnant Women Nonpregnant
Dose/
Sampling
GMTg
GMTg
GMRh
Assay
Time Pointb nf
(95% CIg)
nf
(95% CIg)
(95% CI)h
SARS-CoV-2
neutralisation
assay - NT50
1900.0
2005.7
0.95
(titre)a
2/1 month
99 (1518.2, 2377.7) 113 (1627.0, 2472.6)
(0.69, 1.30)
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre; LLOQ = lower limit
of quantitation; LS = least square; N-binding = SARS-CoV-2 nucleoprotein–binding; NAAT = nucleic acid amplification
test; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
Note: Participants from Study 2 are a selected subset of age matched nonpregnant female Phase 3 participants.
* Participants who had no serological or virological evidence (prior to the 1 month after Dose 2 blood sample collection)
of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Dose 1 and 1 month after Dose 2 and no
positive result between visits, negative NAAT [nasal swab] at Dose 1, Dose 2, and any unscheduled visit prior to the 1
month after Dose 2 blood sample collection) were included in the analysis.
a. SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (original strain [USA-WA1/2020,
isolated in January 2020]).
b. Protocol-specified timing for blood sample collection.
c. n = Number of participants with valid and determinate assay results for the specified assay at the given dose/sampling
time point.
d. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the corresponding
CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 × LLOQ.
e. GMR and 2-sided 95% CIs were calculated by exponentiating the mean difference of the logarithms of the assay and
the corresponding CIs (based on the Student t distribution).
f. n = Number of participants with valid and determinate assay results for the specified assay at both baseline and the
given dose/sampling time point.
g. GMTs and 2-sided CIs were calculated by exponentiating the LS means and the corresponding CIs based on analysis of
log-transformed NT50 titres using a regression model with group, age at Dose 1 in years (continuous), and baseline
log-transformed NT50 titres.
h. GMR (ratio of GMTs of pregnant women to nonpregnant women) and 2-sided CIs were calculated by exponentiating
the difference of LS means and the corresponding CIs based on the same regression model as above.
Among participants without prior evidence of SARS-CoV-2 infection up to 1 month after
Dose 2 (evaluable immunogenicity population), the ratio of the neutralising GMTs (GMR) in
Study C4591015 maternal participants in the BNT162b2 (30 μg) group to that of Study
C4591001 nonpregnant females who received BNT162b2 30 μg was 0.67 (95% CI: 0.50,
0.90).
For participants with or without prior evidence of SARS-CoV-2 infection up to 1 month after
Dose 2 (evaluable immunogenicity population), the model-adjusted ratio of the neutralising
GMTs (adjusted GMR) in Study C4591015 maternal participants in the BNT162b2 (30 μg)
group to that of Study C4591001 nonpregnant females who received BNT162b2 30 μg was
0.95 (95% CI: 0.69, 1.30). The model-adjusted GMT and GMR were calculated based on a
regression model adjusting for age and baseline neutralising titres.
In an additional descriptive immunogenicity analysis, infants born to maternal participants who
received COMIRNATY (tozinameran) had higher geometric mean concentrations (GMCs) of
full length S-binding immunoglobulin G (IgG) concentrations at birth and at 6 months after
delivery [5576.4 (95% CI: 4246.2, 7323.2); n=91 and 311.1 (95% CI: 235.8, 410.5); n=83],
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 25 of 34
respectively, compared to infants born to maternal participants from the placebo group [19.4
(95% CI: 10.2, 37.0); n=92 and 22.0 (95% CI: 11.4, 42.7); n=69].
Immunogenicity in immunocompromised participants (adults and children)
Study C4591024 is a Phase 2b, open-label study (n=124) that enrolled immunocompromised
participants 2 to 17 years of age receiving immunomodulator therapy or who have undergone
solid organ transplant (within the previous 3 months) and are on immunosuppression or who
have undergone bone marrow or stem cell transplant at least 6 months prior to enrollment.
Study C4591024 also enrolled immunocompromised participants 18 years of age and older
treated for NSCLC or CLL, receiving hemodialysis for secondary to end-stage renal disease,
or receiving immunomodulator therapy for an autoimmune inflammatory disorder. Study
participants did not have a past clinical or microbiological diagnosis of COVID-19. Participants
received 4 age-appropriate doses of Comirnaty (tozinameran) (3 micrograms, 10 micrograms,
or 30 micrograms); the first 2 doses separated by 21 days, with the third dose occurring 28 days
after the second dose, followed by a fourth dose, 3 to 6 months after Dose 3.
The immunogenicity results pre-vaccination and after 3 and 4 doses of Comirnaty
(tozinameran) in immunocompromised participants 2 years of age and older are presented in
Table 13.
Table 13. Summary of Geometric Mean Titres – Participants With or Without
Evidence of Infection by Age Group – All-Available Immunogenicity
Population
Comirnaty (tozinameran)
3 micrograms
10 micrograms
30 micrograms 30 micrograms
Age Group:
Age Group:
Age Group:
Age Group:
2 to 4 Years
5 to 11 Years
12 to 17 Years
≥
18 Years
Dose/
Sampling
GMTc
GMTc
GMTc
GMTc
Assay
Time Pointb nc
(95% CId)
nc (95% CId) nc
(95% CId) nc (95% CId)
SARS-CoV-2
44.8
44.5
54.2
82.2
neutralisation
1/Prevax
32
(42.2, 47.7)
62 (42.5, 46.5) 14 (33.7, 87.0) 6 (16.0, 422.5)
assay –
1566.5
2940.6
787.1
reference strain
942.3
(1019.9,
(1175.5,
(66.5,
– NT50 (titre)a 3/1 Month 32 (537.1, 1653.4) 60
2405.9)
14
7356.0)
6
9321.5)
922.2
3284.5
606.2
487.8
(586.7,
(1609.8,
(5.3,
4/Pre-Dose 4 29
(269.0, 884.9) 57
1449.3)
11
6701.3)
3
68756.0)
6463.4
13457.1
1031.3
3447.0
(4319.7,
(5270.1,
(56.9,
4/1 Month 26 (1851.0, 6419.2) 50
9670.9)
9
34362.4)
4
18681.7)
2382.3
5776.1
1605.6
1296.7
(1554.3,
(2801.4,
(28.5,
4/6 Months 25 (674.2, 2494.0) 49
3651.2)
8
11909.2)
3
90614.9)
Abbreviations: CI = confidence interval; GMT = geometric mean titre; LLOQ = lower limit of quantitation; NT50 = 50%
neutralising titre; Prevax = before vaccination; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
a. SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (original strain [USA-WA1/2020, isolated
in January 2020]).
b Protocol-specified timing for blood sample collection.
c. n = Number of participants with valid and determinate assay results for the specified assay at the given dose/sampling time
point.
d. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the corresponding CIs
(based on the Student t distribution). Assay results below the LLOQ were set to 0.5 × LLOQ.
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 26 of 34
Analysis of immunogenicity data at 1 month after Dose 3 (32 participants 2 to 4 years of age,
60 participants 5 to 11 years of age, 14 participants 12 to 17 years of age, and 6 participants
18 years of age and older) and 1 month after Dose 4 (26 participants 2 to 4 years of age, 50
participants 5 to 11 years of age, 9 participants 12 to 17 years of age, and 4 participants 18
years of age and older) in the all available immunogenicity population with or without evidence
of prior infection demonstrated a vaccine-elicited immune response.
GMTs were observed to be substantially higher at 1 month after Dose 3 and further increased
at 1 month after Dose 4 and remained high at 6 months after Dose 4 compared to levels
observed before study vaccination across age groups and disease subsets.
Concomitant vaccine administration with influenza vaccine
In Study C4591030, a Phase 3 multicentre, randomised, observer-blind study, 1,134
participants 18 to 64 years of age who had received 3 doses of Comirnaty (tozinameran) at least
3 months prior were randomised in a 1:1 ratio to receive either Comirnaty (tozinameran)
coadministered with a SIIV, quadrivalent (Afluria Quad) followed 1 month later by placebo
(Group 1, n=568) or an inactivated influenza vaccine with placebo followed 1 month later with
Comirnaty (tozinameran) (Group 2, n=566).
The immune responses to Comirnaty (tozinameran) and SIIV were similar after Comirnaty
(tozinameran) administered concomitantly with SIIV compared with those elicited by either
vaccine administered alone. The non-inferiority criterion was achieved for both full-length S-
binding immunoglobulin G (IgG) and all 4 influenza strain-specific hemagglutination
inhibition (HAI) titres.
The immunogenicity results are presented in Table 14 and Table 15.
Table 14. Geometric Mean Ratio for Full-Length S-Binding IgG Levels (U/mL) at 1
Month After Comirnaty (tozinameran) Vaccination – Evaluable
Immunogenicity Population
Vaccine Group (as Randomised)
Coadministration
Group/Separate
Separate-Administration
Administration
Coadministration Group
Group
Group
GMCb
GMCb
GMRc
Assay
na
(95% CIb)
na
(95% CIb)
(95% CIc)
Full-length
S-binding IgG
13806.5
16254.6
0.83
(U/mL)
499
(12838.9, 14847.0) 413 (15035.5, 17572.5)
(0.77, 0.89)
Abbreviations: CI = confidence interval; GMC = geometric mean concentration; GMR = geometric mean ratio;
IgG = immunoglobulin G; LLOQ = lower limit of quantitation; LS Means = least squares means; S = spike protein.
Note: The baseline was defined as Visit 1 for the coadministration group and Visit 2 for the separate-administration
group.
a. n = Number of participants with valid and determinate assay results for the specified assay at the given
sampling time point.
b. GMC and the 2-sided 95% CI were calculated by exponentiating the concentrations and the corresponding CIs
(based on the Student t distribution). Assay results below the LLOQ were set to 0.5 × LLOQ.
c. GMR and the corresponding 2-sided 95% CI were calculated by exponentiating the difference in LS Means and
the corresponding CIs based on analysis of logarithmically transformed assay results using a linear regression
model with terms of vaccine group, age group, and the corresponding baseline assay results (log scale).
Noninferiority is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67.
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 27 of 34
Table 15. Geometric Mean Ratio for Strain-Specific HAI Titres at 1 Month After SIIV
Vaccination – Evaluable SIIV Immunogenicity Population
Vaccine Group (as Randomised)
Coadministration
Group/Separate
Separate Administration
Administration
Coadministration Group
Group
Group
GMTb
GMTb
GMRc
Strain
na
(95% CIb)
na
(95% CIb)
(95% CIc)
72.4
78.3
0.89
B/Austria
514
(64.2, 81.7)
491
(69.3, 88.5)
(0.77, 1.04)
87.4
86.3
1.00
B/Phuket
520
(79.7, 95.7)
496
(78.4, 94.9)
(0.89, 1.13)
H1N1
344.3
362.3
0.95
A/Victoria
516
(312.4, 379.3)
492
(326.3, 401.6)
(0.83, 1.09)
H3N2
230.6
242.2
0.96
A/Darwin
519
(209.5, 253.8)
491
(221.2, 265.2)
(0.85, 1.09)
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre; HAI =
hemagglutination inhibition; LLOQ = lower limit of quantitation; LS Means = least squares means; SIIV = seasonal
inactivated influenza vaccine; ULOQ = upper limit of quantitation.
Note: The baseline for the SIIV assay was defined at Visit 1.
a. n = Number of participants with valid and determinate assay results for the specified assay at the given
sampling time point.
b. GMTs and the 2-sided 95% CIs were calculated by exponentiating the titres and the corresponding CIs (based
on the Student t distribution). Assay results below the LLOQ were set to 0.5 × LLOQ, and results above the
ULOQ were set to ULOQ + 1.
c. GMRs and the corresponding 2-sided 95% CI were calculated by exponentiating the difference in LS Means
and the corresponding CIs based on analysis of logarithmically transformed assay results using a linear
regression model with terms of vaccine group, age group, and the corresponding baseline assay results (log
scale). Noninferiority is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67.
Concomitant administration with pneumococcal conjugate vaccine
In Study B7471026, a double-blind, randomised descriptive study, participants 65 years of age
and older who had received 2 doses of Comirnaty (tozinameran)
at least 6 months earlier, were
randomised in a 1:1:1 ratio to receive either 20vPnC concomitantly administered with a booster
dose of Comirnaty (tozinameran)
(n=190), or 20vPnC vaccine administered alone (n=191), or
a booster dose of Comirnaty (tozinameran)
administered alone (n=189).
Immune responses to both vaccines were observed after concomitant administration of 20vPnC
vaccine and Comirnaty (tozinameran). Opsonophagocytic activity (OPA) GMTs for the
20 pneumococcal serotypes were similar to 20vPnC vaccine administered alone and IgG
GMCs for the full-length S−binding protein were similar to Comirnaty (tozinameran)
administered alone. A post-hoc analysis found the immune responses to all 20 serotypes
elicited by 20vPnC vaccine when concomitantly administered with Comirnaty (tozinameran)
would have met conventional 2-fold noninferiority criteria compared to 20vPnC vaccine alone,
and the full-length S-binding IgG GMC elicited by Comirnaty (tozinameran)
would have met
conventional 1.5-fold noninferiority criteria compared to Comirnaty (tozinameran)
alone.
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 28 of 34
Concomitant administration with an RSV vaccine or with an RSV vaccine and a high
dose influenza vaccine
In Study C5481001, a Phase 1/2, randomised, multicentre, parallel group, observer-blinded
study 1,083 participants 65 years of age and older who had previously received at least 3 prior
doses of an mRNA COVID-19 vaccine, had not previously received any RSV vaccine, or an
influenza vaccine in the ≤120 days prior to enrolment, were randomised in 1 of 2 enrolment
strata.
The first stratum of approximately 750 participants were randomised 1:1 to evaluate the safety,
tolerability, and immunogenicity of admixed Comirnaty Original/Omicron BA.4-5
(tozinameran/famtozinameran) and RSV (bivalent, recombinant) vaccine concomitantly
administered with high dose quadrivalent flu vaccine or placebo in the opposite arm, compared
to the individual vaccines.
In the second stratum (total participants n=316) participants were randomised 1:1 to receive
Comirnaty Original/Omicron BA.4-5 with concomitantly administered RSV (bivalent,
recombinant) vaccine (in one arm) with either placebo or high dose quadrivalent flu vaccine
(opposite arm). The study objectives included assessing the impact on the immune response of
Comirnaty Original/Omicron BA.4-5 concomitantly administered with RSV (bivalent,
recombinant) vaccine, the immune response of concomitant use of RSV (bivalent,
recombinant) vaccine, Comirnaty Original/Omicron BA.4-5, and high dose quadrivalent flu
vaccine.
When Comirnaty Original/Omicron BA.4-5 was concomitantly administered with RSV
(bivalent, recombinant) vaccine immunologic noninferiority was demonstrated for Comirnaty
Original/Omicron BA.4-5 and RSV (bivalent, recombinant) vaccine compared to individual
administration. The lower limit of the 2-sided 97.5% CI for the GMR for RSV A, RSV B, both
SARS−CoV−2 Omicron BA.4/BA.5 strain and SARS−COV−2 Wuhan-Hu-1 strain (wildtype)
reference strain neutralising titres (NTs) all met the predefined 2-fold noninferiority criterion.
When Comirnaty Original/Omicron BA.4-5 and RSV (bivalent, recombinant) vaccine were
concomitantly administered with high dose quadrivalent flu vaccine, immunologic
noninferiority was demonstrated for Comirnaty Original/Omicron BA.4-5, RSV (bivalent,
recombinant) vaccine and high dose quadrivalent flu vaccine group compared to each
individual administration. The lower limit of the 2-sided 97.5% CI for the GMR for RSV A,
RSV B, both SARS−CoV−2 Omicron BA.4/BA.5 strain and SARS−COV−2 Wuhan-Hu-1
strain (wildtype) reference strain NTs, and each of the 4 strain specific HAI titres all met the
predefined 2-fold noninferiority criterion.
5.2 Pharmacokinetic properties
Not applicable.
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 29 of 34
5.3 Preclinical safety data
Genotoxicity/Carcinogenicity
Neither genotoxicity nor carcinogenicity studies were performed. The components of
Comirnaty (lipids and mRNA) are not expected to have genotoxic potential.
6. PHARMACEUTICAL PARTICULARS
6.1 List of excipients
((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC)
Cholesterol
Trometamol
Trometamol hydrochloride
Sucrose
Water for injections
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those
mentioned in Section 6.6 Special precautions for disposal and other handling.
6.3 Shelf life
Unopened vial
Frozen vial
18 months when stored at -90°C to -60°C.
The vaccine will be received frozen at -90°C to -60°C. Frozen vaccine can be stored either
at -90°C to -60°C or 2°C to 8°C upon receipt.
For thawing instructions of the frozen vials, see Section 6.6 Special precautions for disposal
and other handling.
Thawed vial
If the vaccine is received at 2°C to 8°C it should be stored at 2°C to 8°C. Once removed from
frozen storage, the unopened vial may be stored refrigerated at 2°C to 8°C for a single period
of up to 10 weeks within the 18 month shelf life.
Upon moving the product to 2°C to 8°C storage, the updated expiry date must be written on
the outer carton and the vaccine should be used or discarded by the updated expiry date. The
original expiry date should be crossed out.
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 30 of 34
Check that the expiry date on the outer carton has been updated to reflect the refrigerated expiry
date and that the original expiry date has been crossed out.
Prior to use, the unopened vials can be stored for up to 12 hours at temperatures between
8ºC to 30ºC.
Thawed vials can be handled in room light conditions.
Once thawed, the vaccine should not be re-frozen.
Opened vial
Chemical and physical in-use stability has been demonstrated for 12 hours at 2ºC to 30ºC.
From a microbiological point of view, unless the method of opening precludes the risks of
microbial contamination, the product should be used immediately after the first puncture. If not
used immediately, in-use storage times and conditions are the responsibility of the user.
Pre-filled syringes
Glass pre-filled syringes
The vaccine will be received and stored at 2 °C to 8 °C (refrigerated only).
12 months when stored at 2°C to 8°C Do not freeze. Prior to use, pre-filled syringes can be
stored for up to 12 hours at temperatures between 8 °C and 30 °C and can be handled in room
light conditions.
6.4 Special precautions for storage
Vials
Store multidose vials a in a freezer at −90 °C to −60 °C.
Glass pre-filled syringes
Store glass pre-filled syringes at 2 °C to 8 °C. DO NOT FREEZE.
Vials and pre-filled syringes
Store the vaccine in the original package in order to protect from light. During storage,
minimise exposure to room light, and avoid exposure to direct sunlight and ultraviolet light.
For storage conditions after thawing and first opening, see Section 6.3 Shelf life.
For additional advice on storing Comirnaty LP.8.1, contact Pfizer New Zealand on 0800 736
363.
6.5 Nature and contents of container
Comirnaty LP.8.1 (Dark Grey cap) 2.25 mL fill volume, 2 mL clear multidose vial (Type I
glass) with a stopper (synthetic bromobutyl rubber) and a Dark Grey flip-off plastic cap with
aluminium seal. Each vial contains 6 doses of 0.3 mL, see Section 6.6 Special precautions for
disposal and other handling.
Pack size: 10 vials
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 31 of 34
Comirnaty LP.8.1 Prefilled Glass Syringe: 1 mL clear glass syringe (Type I glass) with
polypropylene rigid cap with a 1 mL plunger stopper (bromobutyl elastomer). Each prefilled
glass syringe contains 1 dose.
Pack size: 10 Prefilled glass syringes
6.6 Special precautions for disposal and other handling
Comirnaty LP.8.1 Suspension for Injection
Handling Instructions for Vaccines in Vials
Handing prior to use
Frozen vials must be completely thawed prior to use. Frozen vials should be transferred to 2 °C
to 8 °C to thaw. Thaw times for 10-vial packs are noted in table below:
Vial Cap Color
Time That May Be Required For a 10-vial Pack
to Thaw (at 2 °C to 8 °C)
Dark Grey
6 hours
• Upon moving frozen vaccine to 2 °C to 8 °C storage, update the expiry date on the carton.
The updated expiry date should reflect 10 weeks from the date of transfer to refrigerated
conditions (2 °C to 8 °C) and not exceeding the original printed expiry date (EXP).
• Alternatively, individual frozen vials may be thawed for 30 minutes at temperatures up to
30 °C for immediate use.
• If the vaccine is received at 2 °C to 8 °C it should continue to be stored at 2 °C to 8 °C.
Check that the carton has been previously updated to reflect the 10-week refrigerated expiry
date.
• Unopened vials can be stored for up to 12 hours at temperatures up to 30 °C. Total storage
time between 8 ºC to 30 ºC, inclusive of storage before and after puncture, should not
exceed 24 hours.
Comirnaty LP.8.1 - Suspension for Injection
Preparation for administration
Comirnaty LP.8.1 Suspension for Injection should be prepared by a healthcare professional
using aseptic technique to ensure the sterility of the prepared suspension.
Vials of Comirnaty LP.8.1 Suspension for Injection have a grey cap, containing6 doses of 0.3
mL of vaccine and
do not require dilution.
o Dark Grey cap: 6 dose multidose vial
Vial verification
Prior to administration, check the name and strength of the vaccine on the vial label and the
colour of the vial cap and vial label border to ensure it is the intended presentation. Check
whether the vial is a single dose vial or a multidose vial and check if the vial requires dilution.
• Check appearance of vaccine prior to mixing and administration.
o Grey cap vials: Prior to mixing, the vaccine is a white to off-white dispersion and
may contain white to off-white opaque amorphous particles.
• Gently invert the vial 10 times.
Do not shake.
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 32 of 34
• Do not use the vaccine if particulates or discoloration are present after mixing.
Preparation of individual doses
• Using aseptic technique, cleanse the vial stopper with a single-use antiseptic swab.
• Withdraw a 0.3 mL single dose.
•
For Dark Grey multidose vials (6 doses per vial):
o After first puncture, record appropriate date and time on the vial and store at 2 ºC
to 30 ºC for up to 12 hours. Do not re-freeze.
o Each dose must contain 0.3 mL of vaccine. Low dead-volume syringes and/or
needles should be used in order to extract all doses from a single vial. The low
dead-volume syringe and needle combination should have a dead volume of no
more than 35 microlitres.
o If the amount of vaccine remaining in the vial cannot provide a full dose, discard
the vial and any excess volume.
Handling Instructions for Vaccines in Prefilled Syringes
Glass pre-filled syringes
• Prior to use, pre-filled syringes can be stored for up to 12 hours at temperatures between 8
°C to 30 °C and can be handled in room light conditions.
Remove tip cap by slowly turning the cap counterclockwise. Do not shake. Attach a needle
appropriate for intramuscular injection and administer the entire volume.
Any unused medicine or waste material should be disposed of in accordance with local
requirements.
7. MEDICINE SCHEDULE
Prescription Medicine.
8. SPONSOR
Pfizer New Zealand Limited
P O Box 3998
Auckland, New Zealand
Toll Free Number: 0800 736 363
9. DATE OF FIRST APPROVAL
Date of publication in the New Zealand Gazette of consent to distribute this medicine:
30 October 2025
10. DATE OF REVISION OF THE TEXT
24 November 2025
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 33 of 34
Comirnaty® is a registered trademark of BioNTech SE. Used under license.
Summary of Updates
Section
Update
4.4
Addition of Study C4591024 data (immunocompromised)
4.5
Addition of unadjuvanted RSV and Pneumococcal conjugate vaccine
coadministration info
4.6
Addition of Study C4591015 data (maternal study)
4.8
Addition of AE data from Study C4591024 & Study C4591015
Addition of data from Study C4591054 SSA & SSB
Addition of unadjuvanted RSV (C5481001) and Pneumococcal
conjugate (B7471026) vaccine coadministration data
4.9
Inclusion of post-authorisation experience
5.1
Addition of Study C4591024 & Study C4591015 clinical data
Addition of data from Study C4591054 SSA & SSB
Addition of unadjuvanted RSV (C5481001) and Pneumococcal
conjugate (B7471026) vaccine coadministration data
Version: pfdclpgi11125
Supersedes: pfdclpgi11025
Page 34 of 34
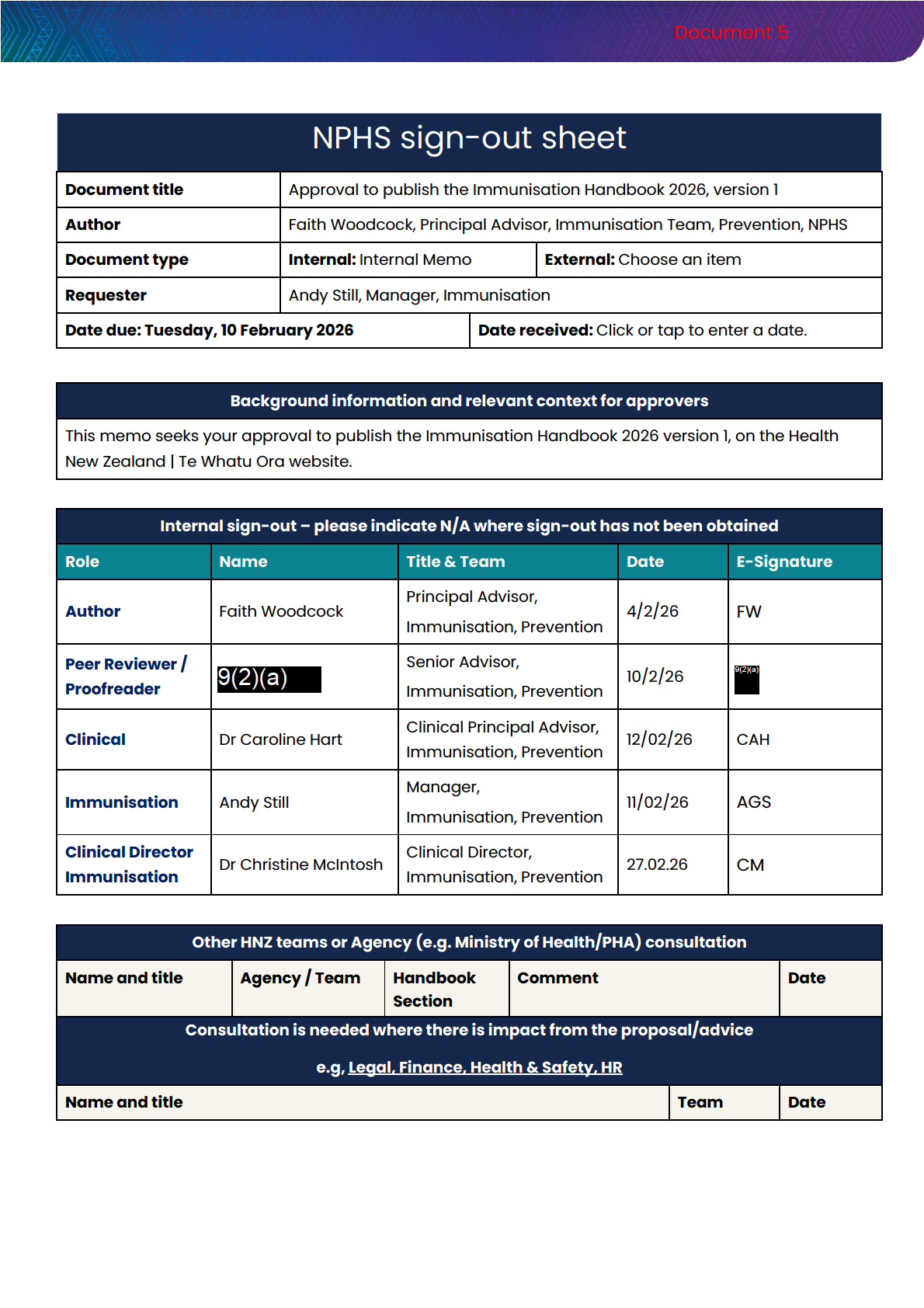




 Handbook updates
Handbook updates
6. Any change to clinical or operational immunisation guidance requires an update to
the Handbook.
7. The Prevention Directorate commissions the Immunisation Advisory Centre (IMAC)
to provide secretariat for the Handbook. IMAC employs a clinical technical writer to
edit the Handbook and contracts clinical expertise from a range of specialties to
provide clinical guidance as the Handbook Advisory Group.
8. The Immunisation team (clinical and operational advisors) within the Prevention
Directorate manage the Handbook update, further reviews as required and the
approval process, through to web publishing on the Health NZ | Te Whatu Ora
website.
9. Final approval to publish is granted by the Clinical Director Immunisation, Prevention,
National Public Health Service.
Immunisation Handbook 2026, version 1
10. The Handbook Advisory Group met on 20 January 2026 to review and agree
changes to the COVID-19 chapter, with the aim of simplifying content and clarifying
dosing recommendations and eligibility criteria for different population groups. The
changes are summarised in
Appendix A.
11. The Handbook has been updated and requires your approval to be published as the
Immunisation Handbook 2026 version 1.
Next steps
12. Once approved, publication will occur as soon as the web version is updated and
relevant IMAC fact sheets are ready.
13. Publication wil be widely communicated to the sector through standard Health NZ |
Te Whatu Ora communications channels including operational stand-up meetings
and the Prevention Pānui (notice) to the sector. Separate communication is shared
with the following:
a. IMAC communications team wil be advised and wil send an email update to
all vaccinators on their mailing list.
b. Protection will be updated to ensure alignment with the Communicable
Disease Control Manual (CD Manual).
c. Pharmac
d. The NZ Formulary team is also updated to ensure the Handbook changes are
reflected in their resource.
Signature: _____________________________________
Date:
Dr Christine McIntosh
Clinical Director Immunisation, Prevention
National Public Health Service
Page | 2
IN-CONFIDENCE

