

133 Molesworth Street
PO Box 5013
Wellington 6140
New Zealand
T+64 4 496 2000
7 November 2024
Steven
By email: [FYI request #28706 email]
Ref:
H2024053438
Tēnā koe Steven
Response to your request for official information
Thank you for your request under the Official Information Act 1982 (the Act) to the Ministry of
Health – Manatū Hauora (the Ministry) on 9 October 2024 for:
“Can you please provide a copy of all submissions made by MTANZ towards the
upcoming Medical device and products legislation”
Six documents have been identified within scope of your request. Al documents are itemised in
Appendix 1 and copies of the documents are enclosed. In determining the release of the
information within these documents, the Ministry of Health consulted with MTANZ. The
documents are being released with no redactions.
I trust this information fulfils your request. If you wish to discuss any aspect of your request with
us, including this decision, please feel free to contact the OIA Services Team
on:
[email address].
Under section 28(3) of the Act, you have the right to ask the Ombudsman to review any
decisions made under this request. The Ombudsman may be contacted by email at:
[email address] or by calling 0800 802 602.
Please note that this response, with your personal details removed, may be published on the
Manatū Hauora website at:
www.health.govt.nz/about-ministry/information-releases/responses-
official-information-act-requests.
Nāku noa, nā
John McGrath
Director Priority Projects
Strategy Policy and Legislation | Te Pou Rautaki
Appendix 1: List of documents for release
# Date
Document details
Decision on release
1 1 March
MTANZ Therapeutic Products Bil
Refused under section 18(d) of the
2023
Submission
Act as the information is publicly
available here:
www.parliament.nz/resource/en-
NZ/53SCHE_EVI_130084_HE4006
5/d0d9623843397b270f7f9e1a9383
d964f95426f2.
2 1 March
MTANZ Therapeutic Products Bil
2023
Submission Supplementary
Refused under section 18(d) of the
Act as the information is publicly
available here:
www.parliament.nz/resource/en-
NZ/53SCHE_EVI_130084_HE4490
0/5ca8102ff4a56e9b9d4f031dcbb44
c20b6561291.
3 June 2024 MTANZ Medical Device Regulatory
Framework Recommendations -
Released in full.
Ministerial Consultation 2June 2024
4 26 July
Letter to Health Select Commit ee –
Refused under section 18(d) of the
2024
MTANZ Submission – Therapeutic
Act as the information is publicly
Products Act Repeal Bill
available here:
www.parliament.nz/resource/en-
NZ/54SCHEA_EVI_8847fc37-a580-
4680-d10f-
08dc93dd1b2f_HEA1563/b8db27c7
d2d3b38778efc45b6b1ee087fddf9e
93.
5 26 August MTANZ Recommendations to Ministry Released in full.
2024
of Health - New Legislation to Replace
the Medicines Act 1981
6 October
Transition Period for Introducing
2024
Medical Device Regulations
Page 2 of 2


Document 3
2024
1982
Act
MTANZ Proposed
Information
Regulatory Framework
Official
for Medica
the l Devices
under
Released
MINISTERIAL CONSULTATION JUNE 2024
CUSHLA SMYTH
Document 3
EXECUTIVE SUMMARY
This report outlines a preferred regulatory framework for medical devices in New Zealand providing
three key recommendations:
1. The need for global harmonisation of medical device legislation and regulations
2. A distinct carve-out provision in the new legislation for medical devices, including IVDs
3. A regulatory recognition model
This proposal aims to ensure a robust regulatory framework for medical devices ensuring patient
safety, fostering innovation, facilitating exports, and streamlining regulatory processes. Adopting
these recommendations wil ensure the availability of safe and effective medical devices and provide
high-quality healthcare solutions to the population.
1982
RECOMMENDATION ONE
‘Ensure the Global Harmonisation of Medical Device Regulations in NZ’
Act
Global harmonisation of regulations for medical devices is critical for several reasons, including
ensuring patient safety, fostering innovation, facilitating exports, and streamlining regulatory
processes. Harmonisation involves aligning regulatory requirements and standards across different
countries to achieve a consistent approach to evaluating and monitoring medical devices.
Key Reasons for Global Harmonisation:
Information
1.
Patient Safety and Public Health:
o
Consistency in Standards: Harmonisation ensures that medical devices meet
consistent safety and performance standards worldwide, reducing the risk of
harmful or substandard products entering the market.
Official
o
Improved Surveil ance: Coordinated post-market surveillance allows for more
the
effective monitoring of adverse events and rapid response to safety concerns,
protecting public health.
2.
Facilitation of Innovation and Market Access:
under
o
Reduced Duplication: Harmonised regulations minimise redundant testing and
documentation requirements, reducing the time and cost for manufacturers to bring
new devices to multiple markets thereby ensuring patient access.
o
Accelerated Innovation: A streamlined regulatory process encourages innovation by
allowing companies to focus resources on research and development rather than
Released
navigating complex regulatory landscapes in different countries.
3.
Economic Benefits:
o
Lower Costs: Harmonisation reduces the financial burden on manufacturers
associated with complying with multiple regulatory systems, leading to cost savings
that can be passed on to healthcare providers and patients.
o
Market Expansion: Enabling access to global markets enhances the commercial
viability of new technologies for NZ innovators, supporting economic growth and job
creation within the medical technology sector.
Document 3
4.
Improved Regulatory Efficiency:
o
Resource Optimisation: Regulatory bodies can allocate resources more effectively
by relying on assessments and approvals conducted by trusted international
counterparts, enhancing overall efficiency.
o
Knowledge Sharing: Harmonisation promotes collaboration and knowledge
exchange among regulators, fostering the adoption of best practices and improving
regulatory capacity.
Jurisdictions Embracing Global Harmonisation:
1.
European Union (EU) - Medical Device Regulation (MDR) and Invitro Diagnostics (IVDR):
o The EU MDR and IVDR aligns with international standards, including those set by the
International Medical Device Regulators Forum (IMDRF) and the Global 1982
Harmonisation Task Force (GHTF). This ensures that devices approved under the
MDR and IVDR meet globally recognised safety and performance criteria.
Act
2.
United States - Food and Drug Administration (FDA):
o The FDA participates in international harmonisation initiatives, such as the IMDRF,
and adopts guidelines that align with international standards. This helps streamline
the approval process for devices intended for both domestic and international
markets.
Information
3.
Australia - Therapeutic Goods Administration (TGA):
o The TGA aligns its regulations with international standards and participates in global
harmonisation efforts through organizations like the IMDRF. This facilitates the
recognition of approvals from other jurisdic
Official tions, simplifying the regulatory process
for manufacturers. the
4.
Singapore - Health Sciences Authority (HSA):
o Singapore’s HSA adopts a regulatory recognition model that acknowledges approvals
from established international regulators. This approach leverages global expertise
under
and ensures that medical devices meet high standards of safety and efficacy.
5.
Canada - Health Canada:
o Health Canada is an active participant in international harmonisation efforts and
aligns its regulations with global standards. This collaboration helps ensure that
medical devices approved in Canada are consistent with those available in other
Released
major markets.
Why Harmonisation is Effective:
•
International Col aboration: Harmonisation efforts, such as those led by the IMDRF,
facilitate collaboration among regulatory authorities, promoting the adoption of best
practices and consistent regulatory frameworks.
•
Regulatory Reliance: Countries like Singapore and Australia use regulatory reliance models,
recognising approvals from trusted international bodies. This approach reduces duplication
of efforts and accelerates market and patient access.
Document 3
•
Standardisation Initiatives: The adoption of international standards, such as ISO 13485 for
quality management systems and ISO 14971 for risk management, ensures a uniform
approach to device safety and performance.
•
Harmonisation and Regulatory Reliance do not impact sovereignty: Pursuing a Harmonised
regulatory reliance model does not prevent New Zealand from making unique decisions to
benefit its public. The Australian TGA has made various decisions tailored to the needs of the
Australian population. Examples include the up classification of surgical mesh devices and
the introduction of Patient Implant Cards (PICs) and Patient Information Leaflets (PILs) for
implantable devices, reflecting early adoption of EU MDR requirements. However, some
regulations, like the reclassification of software-based medical devices, remain specific to
Australia.
‘Key aspects of the Therapeutic Products Act and the 2019 exposure draft were not harmonised
1982
with relevant jurisdictions or IMDRF’
There are many examples of the TPA and the 2018/19 exposure draft not being harmonised, the
Act
most significant being the definition of a medical device itself. Global harmonisation of medical
device regulations is essential for enhancing patient safety, promoting innovation, facilitating
international trade and exports, and improving regulatory efficiency. Jurisdictions like the EU, USA,
Australia, Singapore, and Canada exemplify successful implementation of harmonised regulatory
frameworks, demonstrating the benefits of coordinated global efforts. By aligning regulations with
international standards and collaborating through organisations like the IMDRF, countries can
ensure the availability of safe, effective, and innovative medical devices worldwide.
Information
RECOMMENDATION TWO
‘Implement a Carve-Out Provision for Medical Devices in the Therapeutic Product Legislation’
Official
1.
Objective:
o To establish a distinct regulatory framework for medical devices that ensures their
the
safety, performance, and efficacy while fostering innovation and timely market
access.
2.
Scope:
o The carve out section ap
under plies exclusively to medical devices, which include IVDs,
instruments, apparatus, appliances, software, implants, reagents, materials, or other
articles intended for medical purposes.
3.
Regulatory Requirements:
o
Classification:
Medical devices shall be classified according to their intended use and
inherent risks, following a risk-based classification system similar to the one
Released used by the TGA and the European MDR/IVDR.
o
Clinical Evaluation and Assessment:
Devices must undergo appropriate clinical evaluations and conformity
assessments appropriate to their classification to demonstrate safety and
performance.
o
Post-Market Surveil ance:
Manufacturers must implement post-market surveil ance systems to
monitor device performance and manage any risks that arise after market
entry.
Document 3
4.
Licensing and Facility Requirements:
o Unlike medicines, medical devices shall not require licenses for manufacturing
facilities and wholesaling activities. The regulatory focus will be on the accredited
quality management systems in place and compliance with internationally
recognised standards.
5.
Harmonisation with International Standards:
o The regulatory framework for medical devices will align with global standards, such
as ISO 13485 for quality management systems, to facilitate international trade and
ensure global best practices are followed.
6.
Exemptions and Simplified Procedures:
o Low-risk devices may be subject to simplified regulatory procedures to promote
innovation and reduce unnecessary burdens on manufacturers and suppliers.
1982
By implementing this carve-out, New Zealand can ensure that its regulation of medical devices is on
par with internationally respected models like those of the TGA and European MDR/IVDR, avoiding
Act
unnecessary regulatory burdens and ensuring NZ remains an attractive market for medical device
manufacturers.
‘The Medical Devices frameworks proposed over the years for have been implemented as an
extension of the medicine legislation.’
This co-mingling of legislation leads to incongruencies and complexities for medical Devices and IVDs
Information
that do not align with global medical device regulation frameworks.
The international trend to separate regulatory legislation for medical devices and medicines has
emerged due to the fundamental differences in their development, risk profiles, and mechanisms of
action. This differentiation of legislative provisions is crucial for ensuring that each category receives
Official
appropriate oversight tailored to its specific requirements, thereby enhancing safety, efficacy, and
innovation.
the
Importance of Separate Legislation:
1.
Tailored Evaluation Processes:
under
o
Medicines: These undergo extensive biochemical testing, with a primary focus on
pharmacodynamics, pharmacokinetics, and clinical trials to ensure safety and
efficacy.
o
Medical Devices: These require evaluations based on mechanical, electronic, or
software performance, alongside biocompatibility assessments for implants or in-
Released
body devices. In the case of IVD’s, they are based on clinical and analytical
evaluations. The pace of innovation is faster, demanding a more flexible regulatory
approach, which can be futureproofed for emerging technologies, particularly in the
Digital Health space.
Document 3
2.
Risk Management:
o Medicines typically have well-defined chemical compositions and predictable
biological effects, whereas medical devices can range from simple bandages to
complex surgical robots, each with distinct risk profiles and safety concerns.
3.
Innovation and Market Access:
o The medical device industry benefits from a flexible regulatory framework that can
quickly adapt to technological advancements, promoting faster market entry and
innovation cycles compared to the more static pharmaceutical sector.
Regulatory Framework Exemplars:
1.
Australian Therapeutic Goods Administration (TGA):
1982
o The TGA has distinct regulations for medical devices under the Therapeutic Goods
(Medical Devices) Regulations (2002), together with a separate Chapter (Chapter 4—
Act
Medical devices) within the Therapeutic Goods Act (1989) and many other medical
device specific pieces of subordinate legislation. This framework aligns with
international standards and is designed to ensure that medical devices meet safety
and performance criteria specific to their category and intended use.
o In contrast, medicines are regulated under the "Therapeutic Goods Act 1989,"
focusing on ensuring the safety, quality, and efficacy of pharmaceutical products
through rigorous clinical testing and post-market surveillance.
Information
2.
European Medical Device and IVD Regulation (MDR/IVDR) :
o The European Union implemented the Medical Device Regulation (MDR) in 2017,
which came into ful effect in 2021 and is c
Official urrently progressing through its transition
period. The EU then implemented the Invitro diagnostics regulation (IVDR) in 2022
which is also in its transition period. Both the MDR and IVDR provide a
the
comprehensive regulatory framework specifical y for medical devices and IVDs,
addressing aspects like clinical evaluation, risk classification, and post-market
surveil ance to ensure high standards of safety and performance.
under
o Pharmaceuticals in the EU are regulated under separate legislation, primarily the
"Directive 2001/83/EC" and the "Regulation (EC) No 726/2004," focusing on the
authorization and monitoring of medicinal products for human use, with a distinct
set of requirements for clinical trials and market approval.
By establishing distinct legislative frameworks, both the TGA and the EU ensure that the unique
Released
characteristics and regulatory needs of medical devices and medicines are appropriately addressed,
promoting patient safety, fostering innovation, and improving access to effective healthcare
solutions.
‘Co-mingling of medicines and medical device legislation in the TPA caused scenarios where
medical devices in New Zealand would have been subject to more stringent requirements than
those enforced by the TGA in Australia’
Since the TGA model is widely regarded as an international exemplar in medical device regulation,
these additional regulatory burdens are unnecessary and counterproductive. For instance, under the
Therapeutic Products Act and the 2018/2019 exposure draft, there are licensing requirements for
Document 3
facilities and the wholesaling of medical devices. Such requirements do not exist under the TGA
legislation, which focuses on ensuring safety and efficacy without imposing excessive regulatory
obligations. This divergence highlights the importance of tailored regulatory frameworks that reflect
the distinct needs of the medical device industry, preventing unwarranted administrative and
financial burdens that could stifle innovation and access to medical technologies.
RECOMMENDATION THREE
‘Adopt a Regulatory Recognition Model for Medical Devices in New Zealand’
Adopting a regulatory recognition model for medical devices in New Zealand offers numerous
advantages. This approach leverages the assessments and approvals of trusted international
regulators, such as the FDA, TGA, and EU MDR/IVDR, to streamline the approval process and ensure
the safety and efficacy of medical devices. Adopting a regulatory reliance program for medical
devices is similar to what the current government is proposing for medicines.
1982
Benefits for New Zealand
Act
1.
Overcoming Capability and Capacity Chal enges:
•
Lack of Experience: New Zealand has not previously had a dedicated regulatory system for
medical devices, leading to gaps in expertise and infrastructure. Leveraging approvals from
well-established regulators helps bridge this gap.
•
Building Capacity: A regulatory recognition model allows New Zealand to build its regulatory
Information
capacity over time while ensuring that immediate needs are met without compromising
safety or efficacy.
2.
Efficiency and Timeliness:
Official
•
Faster Approvals: By recognising the certifications from international regulators, New
Zealand can significantly reduce the time required to approve medical devices, ensuring
the
quicker access to new technologies for healthcare providers and patients.
•
Reduced Administrative Burden: The model minimises redundant evaluations and
paperwork, streamlining the regulatory process and making it more efficient.
under
3.
Ensuring High Standards:
•
Global Best Practices: Accepting approvals from reputable regulators ensures that medical
devices in New Zealand meet high international standards of safety, performance, and
quality.
•
Consistency: This approach provides a consistent and reliable framework for manufacturers
Released
and healthcare providers, fostering trust and confidence in the regulatory system.
4.
Economic and Market Benefits:
•
Attracting Innovation: A streamlined and efficient regulatory process makes New Zealand
an attractive market for both global and local medical device manufacturers, promoting
innovation and investment in the medical technology sector.
•
Supporting Local Industry: By reducing regulatory burdens, local manufacturers can bring
their products to market more quickly and compete effectively on a global scale.
Document 3
5. Leveraging Global Expertise:
•
Draw on global knowledge - A regulatory reliance model enables New Zealand to draw on
the extensive global expertise of well-established regulators in regions such as Europe,
Australia, the USA, Japan, Canada, and Singapore. This col aborative approach is common in
jurisdictions where shared knowledge facilitates timely review of premarket assessments.
•
Cyber-security and Artificial Intel igence (AI) – One area where we can benefit from
additional expertise is in the domains of cybersecurity and AI in medical devices. Other
jurisdictions have more advanced legislation and standards in these areas, most notably
Singapore and the UK in areas of cybersecurity, and Europe recently passed the legislation on
AI. By utilising regulatory reliance, we can access this expertise to ensure that these products
are safe and fit for use in our marketplace. This approach allows for the review of complex
issues that require specialised knowledge, which may not be readily available in New
Zealand.
1982
Regulatory Recognition Exemplar - Singapore:
The Health Sciences Authority (HSA) of Singapore uses a regulatory recognition model t
Act hat
acknowledges approvals from reputable international regulators. This model allows the HSA to
expedite the registration process for medical devices by relying on the evaluations and certifications
conducted by trusted regulatory bodies, such as the FDA, TGA, and European MDR/IVDR. Leveraging
a regulatory recognition model has enabled the Singapore HSA to develop into an internationally
respected and established regulatory body, which is now recognised as a reference country by the
TGA.
Information
Success Factors:
1.
Efficiency: By recognising and leveraging the approvals from established regulators,
Singapore's HSA can significantly reduce the time required to bring medical devices to
market.
Official
2.
Resource Optimisation: The HSA can focus its resources on high-risk or novel devices that
may require additional scrutiny, ensu
the ring optimal allocation of regulatory efforts.
3.
Global Standards: Ensuring that medical devices meet international standards by accepting
approvals from trusted regulatory bodies guarantees a high level of safety and efficacy.
4.
Market Competitiveness: The streamlined approval process makes Singapore an attractive
market for medical device man
under ufacturers, encouraging innovation and investment in the
region.
Adopting a regulatory recognition model for medical devices in New Zealand offers a pragmatic and
effective solution to the chal enges posed by the lack of existing regulatory infrastructure and
experience. By leveraging the expertise and approvals of trusted international regulators, New
Zealand can ensure the tim
Released ely availability of safe and effective medical devices, support the growth
of its medical technology industry, and provide high-quality healthcare solutions to its population
whilst still maintaining the sovereignty of decisions made for the benefit of the New Zealand public.
With this model in place, we anticipate that the NZ regulator wil promptly verify the accuracy of the
conformity certificates and subsequently issue an approval to the product sponsor, permitting
importation, local supply and, when needed, exportation. There should be a publicly accessible
database of medical device approvals, providing information on the types of products available in the
market and their respective sponsors. This visibility is crucial for post-market activities, such as
tracing adverse events. It will enable the NZ regulator to concentrate more on post-market
Document 3
monitoring, including the review of real-world evidence, clinical signalling, and continuous
evaluation.
1982
Act
Information
Official
the
under
Released
Document 5
MTANZ RECOMMENDATIONS to MINISTRY OF HEALTH
New Legislation to Replace the Medicines Act 1981
26 August 2024
MTANZ propose the fol owing recommendations for the new legislation to replace the Medicines Act
1981.
1. Define ‘Therapeutic Use’ in the definitions section of the proposed Act and align with the TGA’s
definition which is:
“Therapeutic Use" means use in or in connection with:
a. preventing, diagnosing, curing or alleviating a disease, ailment, defect or injury in
persons; or
b. influencing, inhibiting or modifying a physiological process in persons; or
c. testing the susceptibility of persons to a disease or ailment; or
1982
d. influencing, control ing or preventing conception in persons; or
e. testing for pregnancy in persons; or
Act
f. the replacement or modification of parts of the anatomy in persons.
2. Have individual definitions for Medical Devices and Invitro Diagnostics (IVDs) as per IMDRF
GHTF/SG1/N071:2012 also in the definition section of the proposed Act:
“Medical Device” means any instrument, apparatus, implement, machine, appliance, implant,
reagent for in vitro use, software, material or other similar or related article, intended by the
manufacturer to be used, alone or in combination, for human beings, for one or more of the
Information
specific medical purpose(s) of:
• diagnosis, prevention, monitoring, treatment or alleviation of disease,
• diagnosis, monitoring, treatment, alleviation of or compensation for an injury,
• investigation, replacement, modification, or support of the anatomy or of a
Official
• physiological process,
• supporting or sustaining life,
the
• control of conception,
• disinfection of medical devices,
• providing information by means of in vitro examination of specimens derived from the
human body; and does not achieve its primary intended action by pharmacological,
under
immunological or metabolic means, in or on the human body, but which may be assisted
in its intended function by such means.
“In Vitro Diagnostic (IVD) medical device” means a medical device, whether used alone or in
combination, intended by the manufacturer for the in-vitro examination of specimens derived
from the human body solely or principally to provide information for diagnostic, monitoring or
Released
compatibility purposes. IVD medical devices include reagents, calibrators, control materials,
specimen receptacles, software, and related instruments or apparatus or other articles and are
used, for example, for the following test purposes: diagnosis, aid to diagnosis, screening,
monitoring, predisposition, prognosis, prediction, determination of physiological status.
3.
“Controlled Activities” - Controlled activities for medical devices and IVDs, such as wholesaling
and supply chain management, should fal under the Manufacturer’s or Sponsor’s responsibility
where appropriate. While we recognise the importance of monitoring risks related to product
integrity (storage) and product traceability (recalls), we do not believe these should be classified
as controlled activities. Instead, they should be required as conditions of registration, as is the
case in Australia.
Document 6
‘Medical Device Transition Period’
Introducing Medical Device Regulations in New Zealand
Written by the MTANZ Regulatory Special Advisory Group – October 2024
Introduction
This report outlines a structured transition period for implementing medical device regulations in New
Zealand focusing on alignment with global standards and the International Medical Device Regulators
Forum (IMDRF) recommendations. The framework includes a grandfathering provision for existing medical
1982
devices and In Vitro Diagnostic (IVD) medical devices (col ectively referred to as “medical devices”) to
ensure supply continuity for New Zealand patients, and a phased transition to mandatory Regulator
Act
notification or approval depending on risk classification.
Transition Period Overview
The transition period for implementing medical device regulations in New Zealand spans nine years,
beginning with a three-year grandfathering phase that al ows existing medical devices on the Web Assisted
Notification of Devices (WAND) database and medical devices currently exempt under Schedule 1 of the
Information
Medicines (Database of Medical Devices) Regulations 2003 (“the Regulations”) but previously supplied in
the New Zealand market to remain on the market, contingent upon a Manufacturer/Sponsor declaration
confirming their safety.
The following six years will see mandatory Regulator approval based on both regulatory reliance and non-
Official
reliance models for non-IVD medical devices, starting with Class D medical devices in years four and five,
extending to Class C in years six and seven, and to Class B in years eight and nine. For IVD medical devices
the
with classifications aligning with the IMDRF, the transition should align with non-IVD medical devices, also
commencing with Class D products. During this period of the transition, lower class medical devices that
are not yet required to undergo mandatory Regulator approval wil continue to be introduced to the
market by way of a Manufacturer/Sponsor declaration.
under
Class A products, due to their low risk, will follow a self-declaration model under the Manufacturer’s QMS
commencing in year 9 of the transition period.
This structured approach aims to ensure device safety, enhance Regulatory oversight, minimise disruption
to current safe device supply, mitigate risk to patients and align New Zealand’s framework with global best
practices. Released
It is Industry’s expectation that the Regulator wil be able to select any medical device for an audit at any
stage through, and after, the transition period based on (to be determined) risk factors.
This strategic approach wil foster a smoother transition and support the long-term sustainability of the
medical device and IVD market in New Zealand.
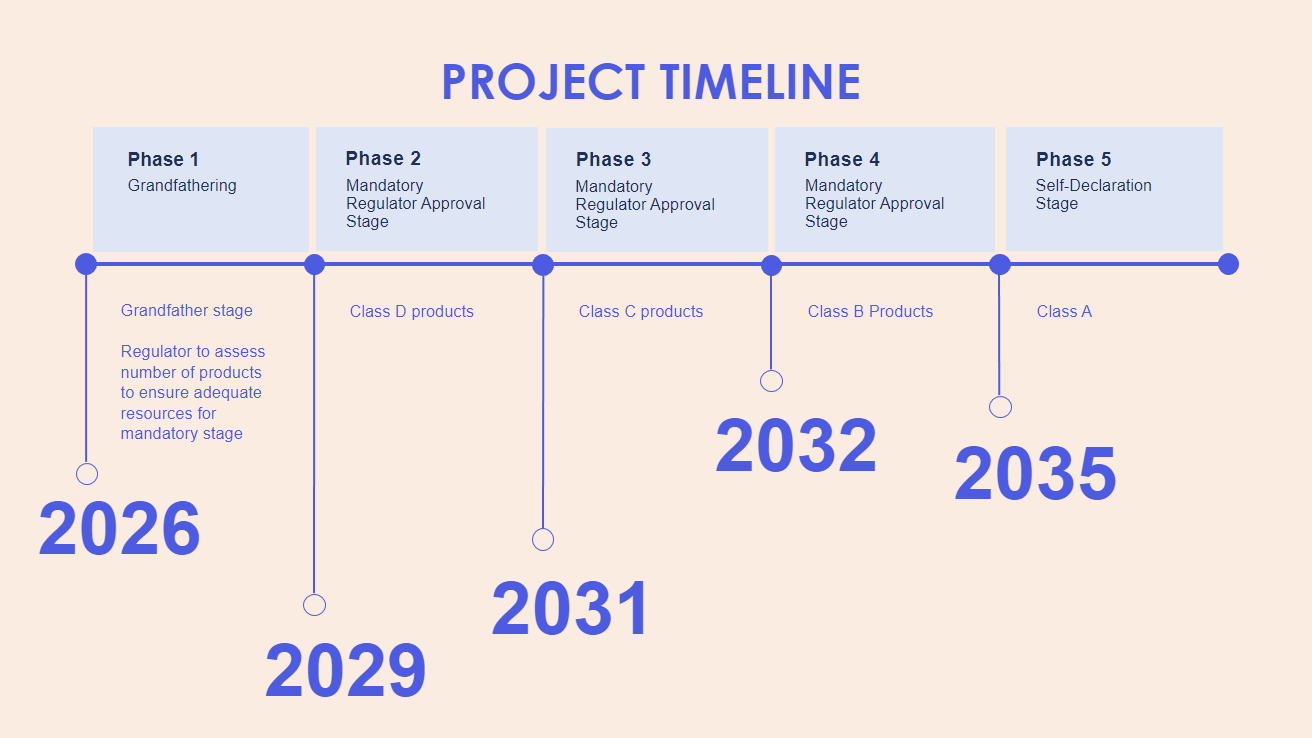
Duration: 9 Years
1
Document 6
Phase 1 Grandfathering Provision
The grandfathering phase will al ow existing medical devices listed on the WAND database and those
currently exempt under Schedule 1 of the Regulations
but previously supplied in the New Zealand market
to remain on the market while providing Sponsors with a structured timeline for compliance.
During the grandfathering stage, Sponsors will notify the Regulator of their intent to continue to supply via
an online portal. Note: Infrastructure wil need to be in place for this portal. Notification wil be made by
grouping devices into similar kinds of devices based on risk classification, GMDN code, and Manufacturer.
The Sponsor must declare that the product:
1982
•
has been supplied within the last 12 months (sold, exported, sampled, donated or for research)
•
is currently listed on WAND or meets the requirements of the exemption under Schedule 1 of the
Act
Regulations
•
complies with a harmonised set of Essential Principles (e.g., IMDRF)
•
is correctly classified
•
has not been requested to be removed from the market in more than one jurisdiction (Australia, EU,
or the USA) due to safety-related issues or other legal infringement(s).
Notifying the Regulator wil offer insight into the devices currently available in New Zealand. This
transparency will provide the Regulator with the advantage of having visibility to the v
Information ariety of devices in
the market, al owing for more effective resource planning and allocation as the mandatory approval or
notification phase approaches. By understanding the volume and types of devices being supplied, the
Regulator can establish the required framework including targeted post-market surveillance and vigilance
activities. This wil also al ow the Regulator to gain a better understanding of resource requirements and
Official
the skil sets those resources wil need to have.
Duration: 3 Years
the
Phase 2-5 Transition to Regulator Approval and Self Declaration
The transition to Regulator approval or no
under tification wil be implemented in phases according to risk
classification.
1. Phase 1 (Year 1-3): Grandfathering provision begins.
2. Phase 2 (Year 4-5): Mandatory Regulator approval begins for Class D
3. Phase 3 (Year 6-7): Mandatory Regulator approval begins for Class C
4. Phase 4 (Year 8-9): Mandatory Regulator approval begins for Class B
Released
5. Phase 5 (Year 9 onwards): Manufacturer Self-Declaration begins for Class A
During the Mandatory approval period, Manufacturers/Sponsors will need to submit documentation by
the reliance or non-reliance models for assessment, as applicable. The Regulator wil approve devices
based on correct documentation being supplied, verification of classification and documentation is current
and meets the stated requirements.
2
Document 6
The regulatory framework should be administered by the Regulator through the development of an online
portal. Sponsors can submit applications to the Regulator through this portal, upload documentation,
make declarations and obtain approval information.
The Regulator should establish legislative timeframes for approvals. Below are the recommended approval
timeframes for each risk class:
• Class A (Lowest Risk): Effective immediately upon self-declaration.
• Class B/C: 20 business days.
• Class D (Highest Risk): 20 business days.
Duration: 6 Years
Risk classification is based on IMDRF classification framework which categorises me
1982 dical
devices and in vitro diagnostics (IVDs) into 4 classes based on their potential risks:
• Class A: Low-risk devices and IVDs (e.g., tongue depressors, simple tests) requiring minimal
Act
regulatory oversight. Also known as Class I in other jurisdictions.
• Class B: Low-Moderate-risk devices and IVDs (e.g., diagnostic ultrasound equipment, tests for non-
critical conditions) needing more comprehensive safety and efficacy data. Also known as Class I a in
other jurisdictions.
• Class C: Moderate-High-risk devices and IVDs (e.g., pacemakers, tests for serious conditions) that
demand extensive clinical data and rigorous scrutiny. Also known as Class I b in other jurisdictions.
• Class D: High-risk devices and IVDs (e.g., implantable devices, tests for life-threatening diseases)
Information
subjected to the most stringent regulatory requirements and extensive evidence. Also known as Class
I I in other jurisdictions
.
Official
Reliance Model (RM)
the
The RM is a process where the Regulator accepts the safety, efficacy, or quality evaluation made by an
approved regulatory authority. This approach aims to streamline regulatory pathways and promote global
harmonisation, minimising duplication of efforts and enabling quicker access to safe and effective medical
devices in various markets. under
Classification
Premarket Requirements
Post Market
Requirements
A
Declaration of Conformance: Sponsors to
Post market obligations for
Low Risk
submit a Manufacturer declaration attesting reporting and field actions.
to compliance with safety and performance
Released
standards.
B
International Registration evidence from a
Post market obligations for
Low-Moderate
recognised regulator. This can include
reporting and field actions.
Risk
registration certificates from Australia,
Canada, EU, Japan, Singapore, USA, or could
also be MDSAP or ISO 13485 Certificate.
3
Document 6
C
International Registration evidence from a
Post market obligations for
Moderate-High
recognised regulator. This can include
reporting and field actions.
Risk
registration certificates from Australia,
Canada, EU, Japan, Singapore, USA.
Three-year reporting
requirement, once approved, to
Manufacturers of implantable devices are
monitor ongoing safety and
also required to submit Patient Information
performance for implantable
Leaflet (PIL) and Patient Information Card
devices.
(PIC), unless specifically excluded from these
requirements.
D
International Registration evidence from a
Post market obligations for
High Risk
recognised regulator. This can include
reporting and field actions.
registration certificates from Australia,
Canada, EU, Japan, Singapore, USA.
Three- year reporting
1982
requirement, once approved, to
Manufacturers of implantable devices are
monitor ongoing safety and
also required to submit Patient Information
performance for implantable
Act
Leaflet (PIL) and Patient Information Card
devices.
(PIC), unless specifically excluded from these
requirements.
Rationale for ISO 13485. Adopting ISO 13485 as a standard is critical
Information for ensuring consistent
quality and safety in medical devices.
ISO 13485 is the standard that specifies requirements for a quality management system. Conformity
assessment is the process of verifying that a standard or technical specification was applied in the
Official
design and manufacturing of a device, which can include compliance with ISO 13485. Generally,
obtaining ISO 13485 certification can be more costly upfront due to the need for documentation,
audits, and potential process changes. Ho
the wever, the cost of conformity assessment varies depending
on the scope and complexity of the device.
under
Non-Reliance Model (NRM)
The NRM pathway is an option where no overseas approval is available. This alternative pathway ensures
that devices can stil be evaluated for safety and efficacy within the local regulatory framework, ensuring
access while maintaining compliance with medical device standards.
Released
Classification Premarket Requirements
Post Market
Requirements
A
Declaration of Conformance: Sponsors to
Post market obligations for
Low Risk
submit a Manufacturer declaration attesting to
reporting and field actions.
compliance with safety and performance
standards.
4
Document 6
B
ISO 13485 Certificate
Post market obligations for
Low-Moderate
reporting and field actions.
Risk
And
Manufacturer Declaration that the device meets
the Essential Principles.
C
ISO 13485 Certificate
Post market obligations for
Moderate-High
reporting and field actions.
Risk
And
Manufacturer Declaration that the device meets Three- year reporting
requirement to monitor
the Essential Principles.
ongoing safety and
Manufacturers of implantable devices are also
performance for implantable
required to submit Patient Information Leaflet
devices.
1982
(PIL) and Patient Information Card (PIC), unless
specifical y excluded from these requirements.
D
ISO 13485 Certificate
Post market obligat
Act ions for
High Risk
reporting and field actions.
And
Manufacturer Declaration that the device meets Three- year reporting
the Essential Principles.
requirement to monitor
ongoing safety and
Manufacturers of implantable devices are also
performance for implantable
required to submit Patient Information Leaflet
devices.
Information
(PIL) and Patient Information Card (PIC), unless
specifical y excluded from these requirements.
*
For certain High-risk devices, the non-reliance
pathway is inappropriate.
Official
the
Recommendations
Priority Recommendations: under
•
Conformity Assessment: Develop New Zealand’s equivalent Essential Principles and conformity
assessment procedures.
•
Monitoring and Evaluation: Establish a framework to assess compliance and effectiveness
throughout the transition period.
•
Online Resources: An online portal would be required to replace WAND and facilitate the
regulatory submissions required. The IT platform should facilitate data retrieval and analytics.
Released
Comprehensive guidance documents, FAQs, and a dedicated help desk should also be established.
This should be a cost-effective effort and ensure that cost is not passed to the sponsor or
manufacturer.
•
Regulatory Statements: Maintain a process al owing Sponsors to request documentation to
support export and registration in overseas jurisdictions, e.g., Certificate of Free Sale or equivalent.
•
International Col aboration: Engage with global Regulatory bodies for knowledge sharing and
alignment with best practices. Consider joining IMDRF as affiliate member.
5
Document 6
•
Post-Implementation Review: Develop evaluation metrics and establish a process for continuous
improvement of the Regulator.
Other Considerations:
•
Communication Strategy: Develop a detailed plan to inform and consult with stakeholders about
new regulations, including timelines and requirements.
•
Pilot Programs: Implement trial runs to gather insights and refine processes before ful -scale
implementation. This could potential y involve a request for Sponsors to volunteer for involvement
in the pilot programs.
•
Stakeholder Engagement: Organise workshops, establish advisory groups, and maintain open lines
of communication with stakeholders.
•
Pre-submission meetings and interactive review consultation meetings between the Regulator
and the Sponsor: Particularly for devices that do not have a clear classification or are new / novel
1982
or are being applied in higher risk environments / patient populations / etc.
•
Financial Support: Explore grants and flexible payment plans to assist smal er NZ Manufacturers.
Act
•
Support Resources: Provide Manufacturers with guidance on ISO 13485 certification processes
and resources for compliance.
•
Technical Assistance: Offer consultation services to help Sponsors/Manufacturers navigate
Regulatory requirements effectively.
Information
Official
the
under
Released
6

Document 6
Project Timeline
1982
Act
Information
Official
the
under
Released
7
Document Outline