NEW ZEALAND DATA SHEET
1. PRODUCT NAME
COMIRNATY® (orange cap, must dilute), new formulation, 0.1 mg/mL concentrate for
suspension for injection, children 5 to 11 years of age (10 micrograms/0.2 mL dose)
Released
2. QUALITATIVE AND QUANTITATIVE COMPOSITION
This is a multidose vial and
must be diluted before use.
One vial (1.3 mL) contains 10 doses of 0.2 mL after dilution, see Section 4.2 Dose and method
of administration and Section 6.6 Special precautions for disposal and other handling.
under the Official Information Act 1982
1 dose (0.2 mL) contains 10 micrograms of tozinameran, a COVID-19 mRNA Vaccine
(embedded in lipid nanoparticles).
Tozinameran is a single-stranded, 5’-capped messenger RNA (mRNA) produced using a cell-
free
in vitro transcription from the corresponding DNA templates, encoding the viral spike (S)
protein of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
For the full list of excipients, see Section 6.1 List of excipients.
3. PHARMACEUTICAL FORM
Concentrate for suspension for injection (sterile concentrate).
COMIRNATY is a white to off-white frozen suspension.
4. CLINICAL PARTICULARS
4.1 Therapeutic indications
COMIRNATY (orange cap, must dilute) has provisional consent (see section 5.1) for the
indication below:
Active immunisation to prevent coronavirus disease 2019 (COVID-19) caused by SARS-CoV-
2, in children aged 5 to 11 years.
The use of this vaccine should be in accordance with official recommendations.
4.2 Dose and method of administration
Dose
Children 5 to 11 years of age (i.e. 5 to less than 12 years of age)
COMIRNATY (orange cap, must dilute) is administered intramuscularly as a primary course
of 2 doses (0.2 mL each) at least 21 days apart.
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 1 of 26
The interchangeability of COMIRNATY with other COVID-19 vaccines to complete the
primary vaccination course has not been established. Individuals who have received 1 dose of
COMIRNATY should receive a second dose of COMIRNATY to complete the primary
vaccination course.
COMIRNATY (orange cap, must dilute) should be used only for children 5 to 11 years of age.
Released
Elderly population
Refer to the Data Sheet for COMIRNATY (grey cap, do not dilute), new formulation, 0.1
mg/mL suspension for injection, 12 years of age and older (30 micrograms/dose).
Method of administration
COMIRNATY should be administered intramuscularly, after dilution. The preferred site of
administration is the deltoid muscle of the upper arm.
under the Official Information Act 1982
Do not inject COMIRNATY intravascularly, subcutaneously or intradermally.
COMIRNATY should not be mixed in the same syringe with any other vaccines or medicinal
products.
For precautions to be taken before administering COMIRNATY, see Section 4.4 Special
warnings and precautions for use.
COMIRNATY (orange cap, must dilute)
Vials have an orange cap and after
dilution contain ten doses of 0.2 mL of vaccine. In order
to extract ten doses from a single vial, low dead-volume syringes and/or needles should be
used. The low dead-volume syringe and needle combination should have a dead volume of no
more than 35 microlitres. If standard syringes and needles are used, there may not be sufficient
volume to extract a tenth dose from a single vial. Irrespective of the type of syringe and needle:
• Each dose must contain 0.2 mL of vaccine.
• If the amount of vaccine remaining in the vial cannot provide a full dose of 0.2 mL, discard
the vial and any excess volume.
• Do not pool excess vaccine from multiple vials.
For instructions on thawing, handling, dilution and dose preparation of COMIRNATY (orange
cap, must dilute) see Section 6.6 Special precautions for disposal and other handling.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of
excipients.
4.4 Special warnings and precautions for use
Traceability
In order to improve the traceability of biological medicinal products, the name and the batch
number of the administered product should be clearly recorded.
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 2 of 26
General recommendations
Hypersensitivity and anaphylaxis
Events of anaphylaxis have been reported. Appropriate medical treatment and supervision
should always be readily available in case of an anaphylactic reaction following the
administration of COMIRNATY.
Released
The individual should be kept under close observation for at least 15 minutes following
vaccination. A second dose of COMIRNATY should not be given to those who have
experienced anaphylaxis to the first dose of COMIRNATY.
Myocarditis and pericarditis
Very rare cases of myocarditis and pericarditis have been observed following vaccination with
COMIRNATY. These cases have primarily occurred within 14 days following vaccination,
more often after the second vaccination, and more often in younger men. Available data
under the Official Information Act 1982
suggest that the course of myocarditis and pericarditis following vaccination is not different
from myocarditis or pericarditis in general.
Healthcare professionals should be alert to the signs and symptoms of myocarditis and
pericarditis. Vaccinees should be instructed to seek immediate medical attention if they
develop symptoms indicative of myocarditis or pericarditis such as (acute and persisting) chest
pain, shortness of breath, or palpitations following vaccination. Healthcare professionals
should consult guidance and/or specialists to diagnose and treat this condition.
Stress-related responses
Some individuals may have stress-related responses associated with the process of vaccination
itself. Stress-related responses are temporary and resolve on their own. They may include
dizziness, fainting, palpitations, increases in heart rate, alterations in blood pressure, feeling
short of breath, tingling sensations, sweating and/or anxiety. Individuals should be advised to
bring symptoms to the attention of the vaccination provider for evaluation and precautions
should be in place to avoid injury from fainting.
Concurrent illness
Vaccination should be postponed in individuals suffering from acute severe febrile illness or
acute infection. The presence of a minor infection and/or low grade fever should not delay
vaccination.
Thrombocytopenia and coagulation disorders
As with other intramuscular injections, COMIRNATY should be given with caution in
individuals receiving anticoagulant ther apy or those with thrombocytopenia or any coagulation
disorder (such as haemophilia) bec ause bleeding or bruising may occur following an
intramuscular administration in these individuals.
Immunocompromised individuals
The efficacy, safety and immunogenicity of COMIRNATY has not been assessed in
immunocompromised individuals, including those receiving immunosuppressant therapy. The
efficacy of COMIRNATY may be lower in immunosuppressed individuals.
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 3 of 26
Duration of protection
The duration of protection afforded by COMIRNATY is unknown as it is still being determined
by ongoing clinical trials.
Limitations of vaccine effectiveness
As with any vaccine, vaccination with COMIRNATY may not protect all vaccine recipients.
Released
Individuals may not be fully protected until 7 days after their second dose of COMIRNATY.
Use in the elderly
Clinical studies of COMIRNATY include participants 65 years of age and older and their data
contributes to the overall assessment of safety and efficacy. See Section 5.1 Pharmacodynamic
properties, Clinical trials, Efficacy against COVID-19.
Paediatric use under the Official Information Act 1982
The safety and efficacy of COMIRNATY in children aged less than 5 years of age have not
yet been established.
Effects on laboratory tests
No data available.
4.5 Interactions with other medicines and other forms of interactions
No interaction studies have been performed.
Concomitant administration of COMIRNATY with other vaccines has not been studied.
4.6 Fertility, pregnancy and lactation
Fertility
In a combined fertility and developmental toxicity study, female rats were intramuscularly
administered COMIRNATY prior to mating and during gestation (4 full human doses of
30 micrograms each, spanning between pre-mating day 21 and gestation day 20). SARS-CoV-
2 neutralising antibodies were present in maternal animals from prior to mating to the end of
the study on postnatal day 21 as well as in fetuses and offspring. There were no vaccine related
effects on female fertility and pregnancy rate.
Pregnancy
There is limited experience with use of COMIRNATY in pregnant women. Animal studies do
not indicate direct or indirect harmful effects with respect to pregnancy, embryo/fetal
development, parturition or post-natal development (see Section 4.6 Fertility, pregnancy and
lactation, Fertility). Administration of COMIRNATY in pregnancy should only be considered
when the potential benefits outweigh any potential risks for the mother and fetus.
Lactation
It is unknown whether tozinameran is excreted in human milk. A combined fertility and
developmental toxicity study in rats did not show harmful effects on offspring development
before weaning (see Section 4.6 Fertility, pregnancy and lactation, Fertility).
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 4 of 26
4.7 Effects on ability to drive and use machines
COMIRNATY has no, or negligible, influence on the ability to drive and use machines.
However, some of the effects mentioned under Section 4.8 Undesirable effects may
temporarily affect the ability to drive or use machines.
4.8 Undesirable effects
Released
Summary of safety profile
The safety of COMIRNATY was evaluated in participants 5 years of age and older in 3 clinical
studies that included 24,675 participants (comprised of 22,026 participants 16 years of age and
older, 1,131 adolescents 12 to 15 years of age and 1,518 children 5 to 11 years of age) that have
received at least one dose of COMIRNATY.
under the Official Information Act 1982
Additionally, 306 existing Phase 3 participants at 18 to 55 years of age received a booster dose
(third dose) of COMIRNATY approximately 6 months after the second dose. The overall
safety profile for the booster dose (third dose) was similar to that seen after 2 doses.
Participants 16 years of age and older – after 2 doses
In Study C4591001, a total of 22,026 participants 16 years of age or older received at least 1
dose of COMIRNATY 30 micrograms and a total of 22,021 participants 16 years of age or
older received placebo (including 138 and 145 adolescents 16 and 17 years of age in the
COMIRNATY and placebo groups, respectively). A total of 20,519 participants 16 years of
age or older received 2 doses of COMIRNATY.
At the time of the analysis of Study C4591001 with a data cut-off of 13 March 2021 for the
placebo-controlled blinded follow-up period up to the participants’ unblinding dates, a total of
25,651 (58.2%) participants (13,031 COMIRNATY and 12,620 placebo) 16 years of age and
older were followed up for ≥4 months after the second dose. This included a total of 15,111
(7,704 COMIRNATY and 7,407 placebo) participants 16 to 55 years of age and a total of
10,540 (5,327 COMIRNATY and 5,213 placebo) participants 56 years and older.
The most frequent adverse reactions in participants 16 years of age and older that received 2
doses were injection site pain (>80%), fatigue (>60%), headache (>50%), myalgia (>40%),
chills (>30%), arthralgia (>20%), pyrexia and injection site swelling (>10%) and were usually
mild or moderate in intensity and resolved within a few days after vaccination. A slightly lower
frequency of reactogenicity events was associated with greater age.
The safety profile in 545 subjects receiving COMIRNATY, that were seropositive for SARS-
CoV-2 at baseline, was similar to that seen in the general population.
Study C4591001 also included 200 participants with confirmed stable human
immunodeficiency virus (HIV) infection. The safety profile of the participants receiving
COMIRNATY (n=100) in the individuals with stable HIV infection was similar to that seen in
the general population.
Adolescents 12 through 15 years of age – after 2 doses
In an analysis of Study C4591001, 2,260 adolescents (1,131 COMIRNATY 30 micrograms;
1,129 placebo) were 12 through 15 years of age. Of these, 1,308 adolescents
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 5 of 26
(660 COMIRNATY and 648 placebo) have been followed for at least 2 months after the second
dose of COMIRNATY. The safety evaluation in Study C4591001 is ongoing.
The most frequent adverse reactions in adolescents 12 through 15 years of age that received 2
doses were injection site pain (>90%), fatigue and headache (>70%), myalgia and chills
(>40%), arthralgia and pyrexia (>20%).
Released
Children 5 to 11 years of age – after 2 doses
In an analysis of Study C4591007 Phase 2/3, 2,268 children (1,518 COMIRNATY
10 micrograms; 750 placebo) were 5 to 11 years of age. Of these, 2,158 (95.1%)
(1,444 COMIRNATY 10 micrograms and 714 placebo) children have been followed for at
least 2 months after the second dose. The safety evaluation in Study C4591007 is ongoing.
The most frequent adverse reactions in children 5 to 11 years of age that received 2 doses
included injection site pain (>80%), fatigue (>50%), headache (>30%), injection site redness
under the Official Information Act 1982
and swelling (>20%), myalgia and chills (>10%).
Participants 18 years of age and older – after booster dose
A subset from Study C4591001 Phase 2/3 participants of 306 adults 18 to 55 years of age who
completed the original COMIRNATY 2-dose course, received a booster dose (third dose) of
COMIRNATY approximately 6 months (range of 4.8 to 8.0 months) after receiving Dose 2.
The most frequent adverse reactions in participants 18 to 55 years of age were injection site
pain (>80%), fatigue (>60%), headache (>40%), myalgia (>30%), chills and arthralgia (>20%).
Tabulated list of adverse reactions from clinical studies and post-authorisation
experience
Adverse reactions observed during clinical studies are listed below according to the following
frequency categories:
Very common (≥ 1/10),
Common (≥ 1/100 to < 1/10),
Uncommon (≥ 1/1,000 to < 1/100),
Rare (≥ 1/10,000 to < 1/1,000),
Very rare (< 1/10,000),
Not known (cannot be estimated from the available data).
Table 1: Adverse reactions from COMIRNATY clinical trials: Individuals 12 years of age
and older
Not known
Rare
Very
Common
Uncommon
(cannot be
System Organ
(≥ 1/10,000
common
(≥ 1/100 to
(≥ 1/1,000 to
estimated from
Class
(≥
to
1/10)
< 1/10)
< 1/100)
the available
< 1/1,000)
data)
Blood and
Lymphadenopathya
lymphatic
system disorders
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 6 of 26
Not known
Rare
Very
Common
Uncommon
(cannot be
System Organ
(≥ 1/10,000
common
(≥ 1/100 to
(≥ 1/1,000 to
estimated from
Class
(≥
to
1/10)
< 1/10)
< 1/100)
the available
< 1/1,000)
data)
Metabolism and
Decreased appetite
nutrition
disorders
Released
Psychiatric
Insomnia
disorders
Nervous system
Headache
Lethargy
Acute
disorders
peripheral
facial
paralysisb
Gastrointestinal
Nausea;
disorders
under the Official Information Act 1982
Skin and
Hyperhidrosis;
subcutaneous
Night sweats
tissue disorders
Musculoskeletal
Arthralgia;
and connective
Myalgia
tissue disorders
General
Injection
Injection
Asthenia; Malaise;
Facial swellingd
disorders and
site pain;
site redness
administration
Fatigue;
site conditions
Chills;
Pyrexiac;
Injection
site
swelling
a A higher frequency of lymphadenopathy (5.2% vs 0.4%) was observed in participants receiving a booster dose
(third dose) compared to participants receiving 2 doses.
b Through the clinical trial safety follow-up period to 14 November 2020, acute peripheral facial paralysis (or
palsy) was reported by four participants in the COMIRNATY group. Onset was Day 37 after Dose 1 (participant
did not receive Dose 2) and Days 3, 9, and 48 after Dose 2. No cases of acute peripheral facial paralysis (or palsy)
were reported in the placebo group.
c A higher frequency of pyrexia was observed after the second dose.
d Facial swelling in vaccine recipients with a history of injection of dermatological fillers
Table 2.
Adverse Reactions from COMIRNATY clinical trial: Individuals 5 to 11
Years of Age (06 September 2021 Data Cut-off Date)
Rare
Common Uncommon
Not known
Very
≥1/100 to ≥1/1,000 to ≥1/10,000
Very Rare
(cannot be
System Organ
Common
to
<1/10
<1/100
<1/10,000
estimated
Class
≥1/10
<1/1,000
(≥1% to
(≥0.1% to
(<0.01%)
from the
(≥10%)
(≥0.01% to
<10%)
<1%)
available data)
<0.1%)
Blood and
Lymphaden
lymphatic system
opathy
disorders
Immune system
Urticariaa,b;
Anaphylaxisa
disorders
Pruritusa,b;
Rasha,b
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 7 of 26
Table 2.
Adverse Reactions from COMIRNATY clinical trial: Individuals 5 to 11
Years of Age (06 September 2021 Data Cut-off Date)
Rare
Common Uncommon
Not known
Very
≥1/100 to ≥1/1,000 to ≥1/10,000
Very Rare
(cannot be
System Organ
Common
to
<1/10
<1/100
<1/10,000
estimated
Class
≥1/10
<1/1,000
(≥1% to
(≥0.1% to
(<0.01%)
from the
(≥10%)
(≥0.01% to
<10%)
<1%)
available data)
Released
<0.1%)
Metabolism and
Decreased
nutrition disorders
appetite
Nervous system
Headache
disorders
Gastrointestinal
Diarrhoeaa; Nausea
disorders
Vomitinga
Musculoskeletal
Myalgia
Arthralgia
Pain in
and connective
extremity
under the Official Information Act 1982
tissue disorders
(arm)a
General disorders
Injection
Pyrexia
Malaise
and administration site pain;
site conditions
Fatigue;
Chills;
Injection
site
swelling;
Injection
site redness
a. These adverse reactions were identified in the post-authorisation period. The following events
were not reported in participants 5 to 11 Years of Age in Study C4591007 but were reported in
individuals ≥16 years of age in Study C4591001: angioedema, lethargy, asthenia, hyperhidrosis,
and night sweats.
b. The following events are categorised as hypersensitivity reactions: urticaria, pruritus, and rash
Post-marketing experience
Although the events listed in Table 3 were not observed in the clinical trials, they are considered
adverse drug reactions for COMIRNATY as they were reported in the post-marketing
experience. As these reactions were derived from spontaneous reports, the frequencies could
not be determined and are thus considered as not known.
Table 3: Adverse reactions from COMIRNATY post marketing experience
System Organ Class
Adverse Drug Reaction
Immune system disorders
Anaphylaxis
Hypersensitivity reactions (e.g. rash, pruritis, urticaria, angioedema)
Cardiac disorders
Myocarditis
Pericarditis
Gastrointestinal disorders
Diarrhoea
Vomiting
Musculoskeletal and connective
Pain in extremity (arm)
tissue disorders
General disorders and
Extensive swelling of vaccinated limb
administration site conditions
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 8 of 26
Reporting suspected adverse effects
Reporting suspected adverse reactions after authorisation of the medicine is important. It allows
continued monitoring of the benefit/risk balance of the medicine. Healthcare professionals are
asked to report any suspected adverse reactions at
https://nzphvc.otago.ac.nz/reporting/.
4.9 Overdose
Released
Overdose data is available from 52 study participants included in the clinical trial that due to
an error in dilution received 58 micrograms of COMIRNATY. The COMIRNATY recipients
did not report an increase in reactogenicity or adverse reactions.
In the event of overdose, monitoring of vital functions and possible symptomatic treatment is
recommended. under the Official Information Act 1982
For advice on the management of overdose please contact the National Poisons Centre on 0800
POISON (0800 764766).
5. PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: vaccines, other viral vaccines, ATC code: J07BX03.
Mechanism of action
The nucleoside-modified messenger RNA in COMIRNATY is formulated in lipid
nanoparticles, which enable delivery of the non-replicating RNA into host cells to direct
transient expression of the SARS-CoV-2 spike (S) antigen. The mRNA codes for membrane-
anchored, full-length S with two point mutations within the central helix. Mutation of these
two amino acids to proline locks S in an antigenically preferred prefusion conformation.
COMIRNATY elicits both neutralising antibody and cellular immune responses to the antigen,
which may contribute to protection against COVID-19.
Clinical efficacy and safety
Efficacy
Study C4591001 is a multicentre, multinational, Phase 1/2/3 randomised, placebo-controlled,
observer-blind dose-finding, vaccine candidate selection and efficacy study in participants 12
years of age and older. Randomisation was stratified by age: 12 to 15 years of age, 16 to 55
years of age, or 56 years of age and older, with a minimum of 40% of participants in the ≥56-
year stratum. The study excluded participants who were immunocompromised and those who
had previous clinical or microbiological diagnosis of COVID-19. Participants with pre-existing
stable disease, defined as disease not requiring significant change in therapy or hospitalisation
for worsening disease during the 6 weeks before enrolment, were included as were participants
with known stable infection with HIV, hepatitis C virus (HCV) or hepatitis B virus (HBV).
Efficacy in participants 16 years of age and older – after 2 doses
In the Phase 2/3 portion of Study C4591001, based on data accrued through
14 November 2020, approximately 44,000 participants were randomised equally and were to
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 9 of 26
receive 2 doses of COMIRNATY or placebo. The efficacy analyses included participants that
received their second vaccination within 19 to 42 days after their first vaccination. The majority
(93.1%) of vaccine recipients received the second dose 19 days to 23 days after Dose 1.
Participants are planned to be followed for up to 24 months after Dose 2, for assessments of
safety and efficacy against COVID-19. In the clinical study, participants were required to
observe a minimum interval of 14 days before and after administration of an influenza vaccine
in order to receive either placebo or COMIRNATY. In the clinical study, participants were
Released
required to observe a minimum interval of 60 days before or after receipt of blood/plasma
products or immunoglobulins through to conclusion of the study in order to receive either
placebo or COMIRNATY.
The population for the analysis of the primary efficacy endpoint included 36,621 participants
12 years of age and older (18,242 in the COMIRNATY group and 18,379 in the placebo group)
who did not have evidence of prior infection with SARS-CoV-2 through 7 days after the second
dose. In addition, 134 participants were between the ages of 16 to 17 years of age (66 in the
under the Official Information Act 1982
COMIRNATY group and 68 in the placebo group) and 1616 participants 75 years of age and
older (804 in the COMIRNATY group and 812 in the placebo group).
At the time of the primary efficacy analysis, participants had been followed for symptomatic
COVID-19 for in total 2,214 person-years for the COMIRNATY group and in total 2,222
person-years for the placebo group.
There were no meaningful clinical differences in overall vaccine efficacy in participants who
were at risk of severe COVID-19 including those with 1 or more comorbidities that increase
the risk of severe COVID-19 (e.g. asthma, body mass index (BMI) ≥30 kg/m2, chronic
pulmonary disease, diabetes mellitus, hypertension).
COMIRNATY efficacy information is presented in Table 4.
Table 4: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2, by age
subgroup – participants without evidence of infection prior to 7 days after Dose 2 –
evaluable efficacy (7 days) population
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Na = 18,198 Cases
Na = 18,325 Cases
Vaccine efficacy
Subgroup
n1b
n1b
%
Surveillance timec
Surveillance timec
(95% CI)f
(n2d)
(n2d)
All participantse
8
162
95.0
2.214 (17,411)
2.222 (17,511)
(90.0, 97.9)
16 to 64 years
7
143
95.1
1.706 (13,549)
1.710 (13,618)
(89.6, 98.1)
65 years and older
1
19
94.7
0.508 (3848)
0.511 (3880)
(66.7, 99.9)
65 to 74 years
1
14
92.9
0.406 (3074)
0.406 (3095)
(53.1, 99.8)
75 years and older
0
5
100.0
0.102 (774)
0.106 (785)
(-13.1, 100.0)
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 10 of 26
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Na = 18,198 Cases
Na = 18,325 Cases
Vaccine efficacy
Subgroup
n1b
n1b
%
Surveillance timec
Surveillance timec
(95% CI)f
(n2d)
(n2d)
Released
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 [*Case definition: (at least 1 of) fever, new or increased cough,
new or increased shortness of breath, chills, new or increased muscle pain, new loss of taste or smell, sore
throat, diarrhoea or vomiting.]
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the last dose) of
past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not
detected by nucleic acid amplification tests (NAAT) [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = number of participants in the specified group.
under the Official Information Act 1982
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of
the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. No confirmed cases were identified in adolescents 12 to 15 years of age.
f. Two-sided confidence interval (CI) for vaccine efficacy (VE) is derived based on the Clopper and Pearson
method adjusted to the surveillance time. CI not adjusted for multiplicity.
In the second primary analysis, efficacy of COMIRNATY in preventing first COVID-19
occurrence from 7 days after Dose 2 compared to placebo was 94.6% (95% credible interval
of 89.9% to 97.3%) in participants 16 years of age and older with or without evidence of prior
infection with SARS-CoV-2.
Additionally, subgroup analyses of the primary efficacy endpoint showed similar efficacy point
estimates across genders, ethnic groups, and participants with medical comorbidities associated
with high risk of severe COVID-19.
Updated efficacy analyses were performed with additional confirmed COVID-19 cases accrued
during blinded placebo-controlled follow-up through 13 March 2021, representing up to
6 months of follow-up after Dose 2 for participants in the efficacy population.
The updated vaccine efficacy information is presented in Table 5.
Table 5: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2, by age
subgroup – participants without evidence of infection prior to 7 days after Dose 2 –
evaluable efficacy (7 days) population during the placebo-controlled follow-up period
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Na=20,998
Na=21,096
Cases
Cases
n1b
n1b
Surveillance Timec
Surveillance Timec
Vaccine efficacy %
Subgroup
(n2d)
(n2d)
(95% CIe)
All participantsf
77
850
91.3
6.247 (20,712)
6.003 (20,713)
(89.0, 93.2)
16 through 64 years
70
710
90.6
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 11 of 26
4.859 (15,519)
4.654 (15,515)
(87.9, 92.7)
65 years and older
7
124
94.5
1.233 (4192)
1.202 (4226)
(88.3, 97.8)
65 through 74 years
6
98
94.1
0.994 (3350)
0.966 (3379)
(86.6, 97.9)
75 years and older
1
26
96.2
0.239 (842)
0.237 (847)
(76.9, 99.9)
Released
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative
at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
under the Official Information Act 1982
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Two-sided confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method
adjusted to the surveillance time.
f. Included confirmed cases in participants 12 through 15 years of age: 0 in the COMIRNATY group (both
without and with or without evidence of prior SARS-CoV-2 infection); 16 and 18 in the placebo group
(without and with or without evidence of prior SARS-CoV-2 infection, respectively).
Efficacy against severe COVID-19 in participants 12 years of age or older – after 2 doses
As of 13 March 2021, vaccine efficacy against severe COVID-19 is presented only for
participants with or without prior SARS-CoV-2 infection (Table 6) as the COVID-19 case
counts in participants without prior SARS-CoV-2 infection were the same as those in
participants with or without prior SARS-CoV-2 infection in both the COMIRNATY and
placebo groups.
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 12 of 26
Table 6. Vaccine Efficacy – First Severe COVID-19 Occurrence in Participants With
or Without* Prior SARS-CoV-2 Infection Based on Food and Drug Administration
(FDA)† Definition After Dose 1 or From 7 Days After Dose 2 in the Placebo-Controlled
Follow-up
COMIRNATY
Placebo
Cases
Cases
Released
n1a
n1a
Vaccine Efficacy %
Surveillance Time (n2b) Surveillance Time (n2b)
(95% CIc)
1
30
96.7
After Dose 1d
8.439e (22,505)
8.288e (22,435)
(80.3, 99.9)
1
21
95.3
7 days after Dose 2f
6.522g (21,649)
6.404g (21,730)
(70.9, 99.9)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at
least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or increased
shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat; diarrhoea;
under the Official Information Act 1982
vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative
at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
† Severe illness from COVID-19 as defined by FDA is confirmed COVID-19 and presence of at least 1 of the
following:
• Clinical signs at rest indicative of severe systemic illness (respiratory rate ≥30 breaths per minute, heart
rate ≥125 beats per minute, saturation of oxygen ≤93% on room air at sea level, or ratio of arterial
oxygen partial pressure to fractional inspired oxygen <300 mm Hg);
• Respiratory failure [defined as needing high-flow oxygen, noninvasive ventilation, mechanical
ventilation or extracorporeal membrane oxygenation (ECMO)];
• Evidence of shock (systolic blood pressure <90 mm Hg, diastolic blood pressure <60 mm Hg, or
requiring vasopressors);
• Significant acute renal, hepatic, or neurologic dysfunction;
• Admission to an Intensive Care Unit;
• Death.
a. n1 = Number of participants meeting the endpoint definition.
b. n2 = Number of participants at risk for the endpoint.
c. Two-side confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method
adjusted to the surveillance time.
d. Efficacy assessed based on the Dose 1 all available efficacy (modified intention-to-treat) population that
included all randomised participants who received at least 1 dose of study intervention.
e. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from Dose 1 to the end of the surveillance
period.
f. Efficacy assessed based on the evaluable efficacy (7 Days) population that included all eligible randomised
participants who receive all dose(s) of study intervention as randomised within the predefined window, have
no other important protocol deviations as determined by the clinician
g. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
Efficacy and immunogenicity in adolescents 12 to 15 years of age – after 2 doses
An analysis of Study C4591001 has been performed in adolescents 12 to 15 years of age up to
a data cutoff date of 13 March 2021.
The vaccine efficacy information in adolescents 12 to 15 years of age is presented in Table 7.
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 13 of 26
Table 7: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2 –
participants without evidence of infection and with or without evidence of infection prior
to 7 days after Dose 2 – adolescents 12 to 15 years of age evaluable efficacy (7 days)
population
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
without evidence of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Released
Na = 1005
Na = 978
Cases
Cases
n1b
n1b
Vaccine efficacy
Surveillance timec (n2d)
Surveillance timec (n2d)
% (95% CIe)
Adolescents
0
16
12 to 15 years
0.154 (1001)
0.147 (972)
100.0 (75.3, 100.0)
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
with or without* evidence of prior SARS-CoV-2 infection
under
COMIRNATY
Placebo
Na = 1119
Na = 1110
Cases
Cases
n1b
n1b
Vaccine efficacy
Surveillance timec (n2d)
Surveillance timec (n2d)
% (95% CIe)
the
Adolescents
0
18
12 to 15 years
0.170 (1109)
0.163 (1094)
100.0 (78.1, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at
Official
least 1 symptom consistent with COVID-19 [*Case definition: (at least 1 of) fever, new or increased cough, new
or increased shortness of breath, chills, new or increased muscle pain, new loss of taste or smell, sore throat,
diarrhoea or vomiting).
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the last dose) of
past SARS-CoV-2 infection (i.e, N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not
detected by nucleic acid amplification tests (NAAT) [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
Information
a. N = number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all subjects within each group at
risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of subjects at risk for the endpoint.
e. Confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method adjusted
for surveillance time. CI not adjusted for multiplicity.
In Study C4591001 an analysis of SARS-CoV-2 neutralising titres in a randomly selected
Act
subset of participants was performed to demonstrate non-inferior immune responses (within
1.5-fold) comparing adolescents 12 to 15 years of age to participants 16 to 25 years of age who
had no serological or virological evidence of past SARS-CoV-2 infection. The immune
1982
response to COMIRNATY in adolescents 12 to 15 years of age (n = 190) was non-inferior to
the immune response in participants 16 to 25 years of age (n = 170), based on results for SARS-
CoV-2 neutralising titres at 1 month after Dose 2. The geometric mean titres (GMT) ratio of
the adolescents 12 to 15 years of age group to the participants 16 to 25 years of age group was
1.76, with a 2-sided 95% CI of 1.47 to 2.10, meeting the 1.5-fold non-inferiority criterion (the
lower bound of the 2-sided 95% CI for the geometric mean ratio [GMR] >0.67), which
indicates a statistically greater response in the adolescents 12 to 15 years of age than that of
participants 16 to 25 years of age.
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 14 of 26
Immunogenicity in children 5 to 11 years of age – after 2 doses
Study C4591007 is a Phase 1/2/3 study comprised of an open-label vaccine dose-finding
portion (Phase 1) and a multicentre, multinational, randomised, saline placebo-controlled,
observer-blind efficacy portion (Phase 2/3) that has enrolled participants 5 through to 11 years
of age.
In C4591007, an analysis of SARS-CoV-2 50% neutralising titres (NT50) 1 month after Dose
Released
2 in a randomly selected subset of participants demonstrated effectiveness by immunobridging
of immune responses comparing children 5 to 11 years of age in the Phase 2/3 part of Study
C4591007 to participants 16 to 25 years of age in the Phase 2/3 part of Study C4591001 who
had no serological or virological evidence of past SARS-CoV-2 infection up to 1 month after
Dose 2, meeting the prespecified immunobridging criteria for both the geometric mean ratio
(GMR) and the seroresponse difference with seroresponse defined as achieving at least 4-fold
rise in SARS-CoV-2 NT50 from baseline (before Dose 1).
under
The ratio of the SARS-CoV-2 NT50 in children 5 to 11 years of age to that of young adults 16
to 25 years of age was 1.04 (2-sided 95% CI: 0.93, 1.18), as presented in Table 8.
Table 8: Summary of geometric mean ratio for 50% neutralising titre – Comparison of
the
children 5 to 11 years of age (Study C4591007) to participants 16 to 25 years of age (Study
C4591001) – participants without* evidence of infection up to 1 month after Dose 2 –
evaluable immunogenicity population
Official
COMIRNATY
5 to 11 years/
10 microgram/dose 30 microgram/dose
16 to 25 years
5 to 11 years
16 to 25 years
na=264
na=253
Met
Time
GMTc
GMTc
GMRd
immunobridging
Information
Assay
pointb
(95% CIc)
(95% CIc)
(95% CId)
objectivee
(Y/N)
SARS-CoV-2
neutralisation 1 month
1197.6
1146.5
1.04
Y
assay - NT50 after
(1106.1, 1296.6)
(1045.5, 1257.2)
(0.93, 1.18)
(titre)f
Dose 2
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre;
LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test; NT50 = 50% neutralising titre;
SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
*Participants who had no serological or virological evidence (up to 1 month post-Dose 2 blood sample collection)
Act
of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and 1 month after Dose
2, SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2, and negative NAAT (nasal swab) at
any unscheduled visit up to 1 month after Dose 2 blood collection) and had no medical history of COVID-
1982
19 were included in the analysis.
a. n = Number of participants with valid and determinate assay results for the specified assay at the given
dose/sampling time point.
b. Protocol-specified timing for blood sample collection.
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
d. GMRs and 2-sided 95% CIs were calculated by exponentiating the mean difference of the logarithms of the
titres (Group 1[5 to 11 years of age] - Group 2 [16 to 25 years of age]) and the corresponding CI (based on
the Student t distribution).
e. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67 and
the point estimate of the GMR is ≥0.8.
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 15 of 26
f. SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation
Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus
neutralisation is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution
at which 50% of the virus is neutralised.
Among participants without prior evidence of SARS-CoV-2 infection up to 1 month after
Dose 2, 99.2% of children 5 to 11 years of age and 99.2% of participants 16 to 25 years of age
had a seroresponse from before vaccination to 1 month after Dose 2. The difference in
Released
proportions of participants who had seroresponse between the 2 age groups (children – young
adult) was 0.0% (2-sided 95% CI: -2.0%, 2.2%) as presented in Table 9.
Table 9:
Difference in percentages of participants with seroresponse – participants
without evidence of infection up to 1 month after Dose 2 – immunobridging subset –
Phase 2/3 – comparison of 5 to 11 years of age to Study C4591001 Phase 2/3 16 to 25 years
of age – evaluable immunogenicity population
COMIRNATY
under
10
30
5 to 11 years/
microgram/dose microgram/dose
16 to 25 years
5 to 11 years
16 to 25 years
Na=264
Na=253
the
Met
Time
nc (%)
nc (%)
Difference %e immunobridging
Assay
pointb
(95% CId)
(95% CId)
(95% CIf)
objectiveg
Official
(Y/N)
SARS-CoV-2
1 month
neutralisation
262 (99.2)
251 (99.2)
0.0
after
Y
assay – NT50
(97.3, 99.9)
(97.2, 99.9)
(-2.0, 2.2)
Dose 2
(titre)h
Abbreviations: LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test;
Information
N-binding = SARS-CoV-2 nucleoprotein–binding; NT50 = 50% neutralising titre 50; SARS-CoV-2 = severe acute
respiratory syndrome coronavirus 2.
Note: Seroresponse is defined as achieving a ≥4-fold rise from baseline (before Dose 1). If the baseline measurement
is below the LLOQ, a postvaccination assay result ≥4 × LLOQ is considered a seroresponse.
Note: Participants who had no serological or virological evidence (up to 1 month post-Dose 2 blood sample
collection) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and 1 month after
Dose 2, SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2, and negative NAAT (nasal swab) at
any unscheduled visit up to 1 month after Dose 2 blood collection) and had no medical history of COVID-19 were
included in the analysis.
a. N = number of participants with valid and determinate assay results both before vaccination and at 1 month after
Dose 2. These values are the denominators for the percentage calculations.
Act
b. Protocol-specified timing for blood sample collection.
c. n = Number of participants with seroresponse for the given assay at the given dose/sampling time point.
d. Exact 2-sided CI based on the Clopper and Pearson method.
e. Difference in proportions, expressed as a percentage (Group 1 [5 to 11 years of age] – Group 2 [16 to 25 years
1982
of age]).
f. 2-Sided CI, based on the Miettinen and Nurminen method for the difference in proportions, expressed as a
percentage.
g. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the difference in proportions is greater
than -10.0%.
h. SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation Assay.
The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus neutralisation is
read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution at which 50% of
the virus is neutralised.
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 16 of 26
Immunogenicity in participants 18 years of age and older – after booster dose
Effectiveness of a booster dose of COMIRNATY was based on an assessment of 50%
neutralising titres (NT50) against SARS-CoV-2 (USA_WA1/2020). In Study C4591001,
analyses of NT50 1 month after the booster dose compared to 1 month after the primary series
in individuals 18 to 55 years of age who had no serological or virological evidence of past
SARS-CoV-2 infection up to 1 month after the booster vaccination demonstrated
noninferiority for both GMR and difference in seroresponse rates. Seroresponse for a
Released
participant was defined as achieving a ≥4-fold rise in NT50 from baseline (before Dose 1),
These analyses are summarised in Table 10.
Table 10. SARS-CoV-2 neutralisation assay - NT50 (titre)† (SARS-CoV-2
USA_WA1/2020) – GMT and seroresponse rate comparison of 1 month after booster dose
to 1 month after primary series – participants 18 to 55 years of age without evidence of
infection up to 1 month after booster dose* – booster dose evaluable immunogenicity
under
population±
1 month after
booster dose/-
1 month after
Met
1 month after
1 month after
primary
noninferiority
the
booster dose
primary series
series
objective
n
(95% CI)
(95% CI)
(97.5% CI)
(Y/N)
Geometric mean
50% neutralising
2466.0b
750.6
b
3.29c
Official
titre (GMTb)
212a
(2202.6, 2760.8)
(656.2, 858.6)
(2.77, 3.90)
Yd
Seroresponse rate
199f
196f
(%) for 50%
99.5%
98.0%
1.5%g
neutralising titre†
200e
(97.2%, 100.0%) (95.0%, 99.5%) (-0.7%, 3.7%
h)
Yi
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre;
LLOQ = lower limit of quantitation; N-binding = SARS-CoV-2 nucleoprotein-binding; NAAT = nucleic acid
Information
amplification test; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute respiratory syndrome
coronavirus 2; Y/N = yes/no.
†
SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation
Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus
neutralization is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum
dilution at which 50% of the virus is neutralised.
*
Participants who had no serological or virological evidence (up to 1 month after receipt of a booster dose
of Comirnaty) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative and
SARS-CoV-2 not detected by NAAT [nasal swab]) and had a negative NAAT (nasal swab) at any
unscheduled visit up to 1 month after the booster dose were included in the analysis.
Act
± All eligible participants who had received 2 doses of Comirnaty as initially randomised, with Dose 2
received within the predefined window (within 19 to 42 days after Dose 1), received a booster dose of
Comirnaty, had at least 1 valid and determinate immunogenicity result after booster dose from a blood
collection within an appropriate window (within 28 to 42 days after the booster dose), and had no other
1982
important protocol deviations as determined by the clinician.
a. n = Number of participants with valid and determinate assay results at both sampling time points within
specified window.
b. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
c. GMRs and 2-sided 97.5% CIs were calculated by exponentiating the mean differences in the logarithms of
the assay and the corresponding CIs (based on the Student t distribution).
d. Noninferiority is declared if the lower bound of the 2-sided 97.5% CI for the GMR is > 0.67 and the point
estimate of the GMR is ≥ 0.80.
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 17 of 26
e. n = Number of participants with valid and determinate assay results for the specified assay at baseline,
1 month after Dose 2 and 1 month after the booster dose within specified window. These values are the
denominators for the percentage calculations.
f. Number of participants with seroresponse for the given assay at the given dose/sampling time point. Exact
2-sided CI based on the Clopper and Pearson method.
g. Difference in proportions, expressed as a percentage (1 month after booster dose – 1 month after Dose 2).
h. Adjusted Wald 2-sided CI for the difference in proportions, expressed as a percentage.
i. Noninferiority is declared if the lower bound of the 2-sided 97.5% CI for the percentage difference is
Released
> -10%.
This medicine has been given a provisional consent under Section 23 of the Act. This means
that further evidence on this medicine is awaited or that there are specific conditions of use.
Refer to the consent notice published in the New Zealand Gazette for the specific conditions.
5.2 Pharmacokinetic properties
under
Not applicable.
5.3 Preclinical safety data
the
Genotoxicity/Carcinogenicity
Neither genotoxicity nor carcinogenicity studies were performed. The components of
COMIRNATY (lipids and mRNA) are not expected to have genotoxic potential.
Official
6. PHARMACEUTICAL PARTICULARS
6.1 List of excipients
Information
((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
Distearoylphosphatidylcholine (DSPC)
Cholesterol
Trometamol
Trometamol hydrochloride
Act
Sucrose
Water for injections
1982
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those
mentioned in Section 6.6 Special precautions for disposal and other handling.
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 18 of 26
6.3 Shelf life
COMIRNATY (orange cap, must dilute)
Unopened vial
Frozen vial
9 months when stored at -90°C to -60°C.
Released
The vaccine will be received frozen at -90°C to -60°C. Frozen vaccine can be stored either at
-90°C to -60°C or 2°C to 8°C upon receipt.
When stored frozen at -90°C to -60°C, 10-vial packs of the vaccine can be thawed at 2°C to 8°C
for 4 hours or individual vials can be thawed at room temperature (up to 30°C) for 30 minutes.
Thawed vial under
If the vaccine is received at 2°C to 8°C it should be stored at 2°C to 8°C. Once removed from
frozen storage, the unopened vial may be stored refrigerated at 2°C to 8°C for a single period
of up to 10 weeks within the 6-month shelf life.
the
Upon moving the product to 2°C to 8°C storage, the updated expiry date must be written on
the outer carton and the vaccine should be used or discarded by the updated expiry date. The
original expiry date should be crossed out.
Official
Check that the expiry date on the outer carton has been updated to reflect the refrigerated expiry
date and that the original expiry date has been crossed out.
When stored frozen at -90°C to -60°C, the vaccine can be thawed at either 2°C to 8°C or at
temperatures up to 30°C.
Information
Prior to use, the unopened vials can be stored for up to 12 hours at temperatures between
8ºC to 30ºC.
Thawed vials can be handled in room light conditions.
Once thawed COMIRNATY (orange cap, must dilute) should not be re-frozen.
Diluted medicinal product
Act
Chemical and physical in-use stability has been demonstrated for 12 hours at 2ºC to 30°C, after
dilution with sodium chloride 9 mg/mL (0.9%) solution for injection. From a microbiological
point of view, unless the method of dilution precludes the risk of microbial contamination, the
1982
product should be used immediately. If not used immediately, in-use storage times and
conditions are the responsibility of the user.
6.4 Special precautions for storage
COMIRNATY (orange cap, must dilute) can be stored in a refrigerator at 2°C to 8°C for a
single period of up to 10 weeks, not exceeding the original expiry date (EXP). The expiry date
for storage at -90°C to -60°C is printed on the vial and outer carton after “EXP”.
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 19 of 26


Check that the expiry date has been updated to reflect the refrigerated EXP date and that the
original expiry date has been crossed out.
Store in the original package to protect from light. During storage, minimise exposure to room
light, and avoid exposure to direct sunlight and ultraviolet light.
For detailed instructions see Section 6.6 Special precautions for disposal and other handling.
Released
Once thawed, the vaccine cannot be re-frozen.
Thawed vials can be handled in room light conditions.
For storage conditions after thawing and dilution of the medicinal product, see Section 6.3
Shelf life.
under
For additional advice on storing COMIRNATY, contact Pfizer New Zealand on 0800 736 363.
6.5 Nature and contents of container
COMIRNATY (orange cap, must dilute) 1.3 mL fill volume in 2 mL clear multidose vial
the
(Type I glass) with a stopper (synthetic bromobutyl rubber) and an orange flip-off plastic cap
with aluminium seal. Each vial contains 10 doses, see Section 6.6 Special precautions for
disposal and other handling.
Official
Pack size: 10 vials, 195 vials
Not all pack sizes may be marketed.
Information
6.6 Special precautions for disposal and other handling
COMIRNATY (orange cap, must dilute)
The vaccine should be prepared by a healthcare professional using aseptic technique to ensure
the sterility of the prepared diluted suspension.
COMIRNATY (orange cap, must dilute)
Dose Verification
Act
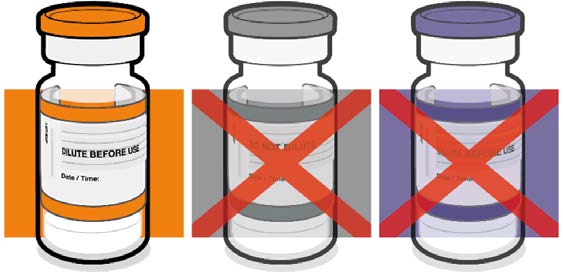
• Verify that the vial has an orange
plastic cap.
Orange cap
1982
• Only the orange cap vial can be used
for children age 5 to 11 years.
10 micrograms
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 20 of 26
 COMIRNATY (orange cap, must dilute)
Handling Prior To Use
COMIRNATY (orange cap, must dilute)
Handling Prior To Use
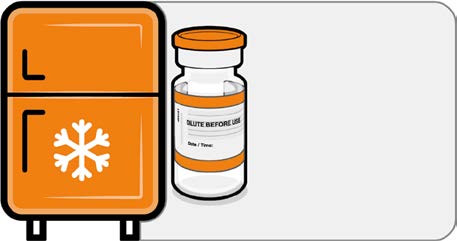
• If the multidose vial is stored frozen
it must be thawed prior to use.
Frozen vials should be transferred to
Released
an environment of 2°C to 8°C to
thaw; a 10 vial pack may take
4 hours to thaw. Ensure vials are
Store for up to
completely thawed prior to use.
10 weeks at
• Unopened vials can be stored for up
2 °C to 8 °C
to 10 weeks at 2°C to 8°C within the
9 month shelf life.
•
under
Alternatively, individual frozen vials
may be thawed for 30 minutes at
temperatures up to 30°C for
immediate use.
the Official
Information
Act 1982
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 21 of 26
 COMIRNATY (orange cap, must dilute)
Mixing Prior To Dilution
COMIRNATY (orange cap, must dilute)
Mixing Prior To Dilution
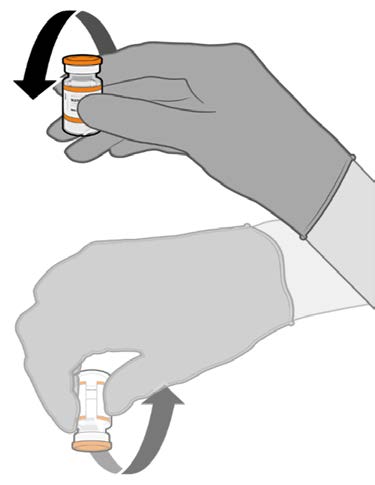
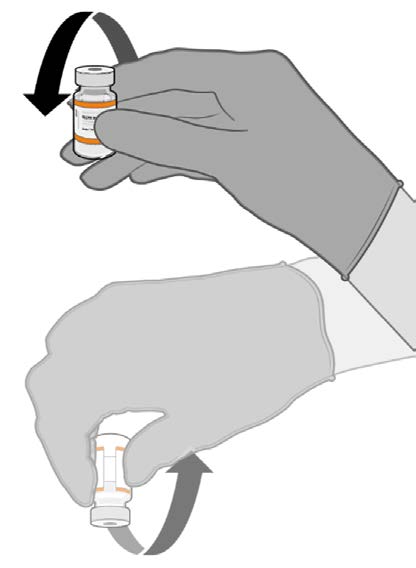
• Allow the thawed vial to come to
room temperature and gently invert it
10 times prior to dilution. Do not
Released
shake.
• Prior to dilution, the thawed
suspension may contain white to off-
white opaque amorphous particles.
under
the
Official
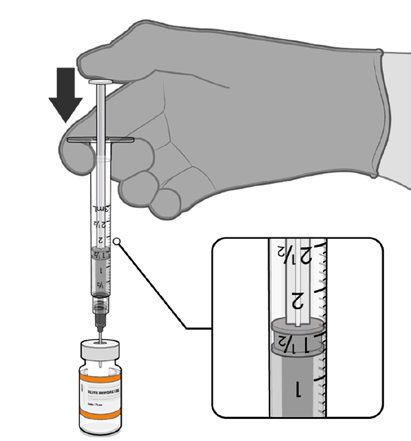
Dilution
• The thawed vaccine must be diluted
in its original vial with 1.3 mL
sodium chloride 9 mg/mL (0.9%)
solution for injection, using a
Information
21 gauge or narrower needle and
aseptic techniques.
Act 1982
1.3 mL of 0.9% sodium chloride
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 22 of 26
 COMIRNATY (orange cap, must dilute)
Dilution (continued)
COMIRNATY (orange cap, must dilute)
Dilution (continued)
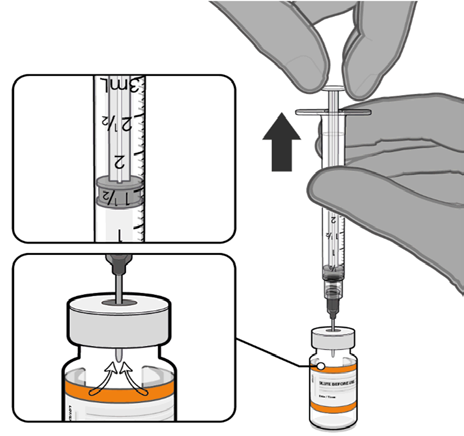
• Equalise vial pressure before
removing the needle from the vial
stopper by withdrawing 1.3 mL air
Released
into the empty diluent syringe.
under
the
Pull back plunger to 1.3 mL to
remove air from vial.
Official
• Gently invert the diluted suspension
10 times. Do not shake.
• The diluted vaccine should present as
a white to off-white suspension with
no particulates visible. Do not use
Information
the diluted vaccine if particulates or
discoloration are present.
Act 1982
Gently × 10
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 23 of 26
 COMIRNATY (orange cap, must dilute)
Dilution (continued)
COMIRNATY (orange cap, must dilute)
Dilution (continued)
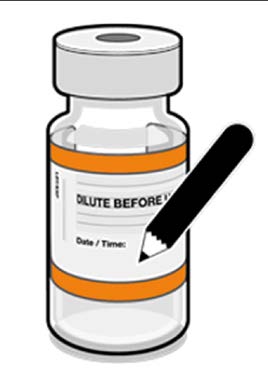
• The diluted vials should be marked
with the appropriate date and time.
• After dilution, store at 2°C to 30°C
Released
and use within 12 hours.
• Do not freeze or shake the diluted
dispersion. If refrigerated, allow the
diluted suspension to come to room
temperature prior to use.
under
Record appropriate date and time.
Use within 12 hours after dilution.
the
Official
Information
Act 1982
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 24 of 26
 COMIRNATY (orange cap, must dilute)
Preparation of Individual 0.2 mL Doses of COMIRNATY (orange cap, must dilute)
COMIRNATY (orange cap, must dilute)
Preparation of Individual 0.2 mL Doses of COMIRNATY (orange cap, must dilute)
• Using aseptic technique, cleanse the
vial stopper with a single-use
antiseptic swab.
Released
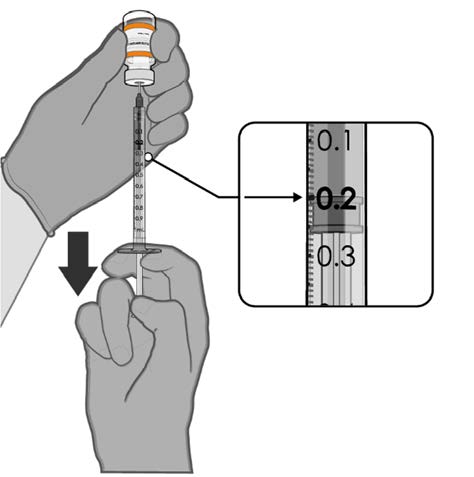
• Withdraw 0.2 mL of COMIRNATY
(orange cap, must dilute).
Low dead-volume syringes and/or
needles should be used in order to
extract 10 doses from a single vial.
The low dead-volume syringe and
needle combination should have a
under
dead volume of no more than
35 microlitres.
If standard syringes and needles are
the
used, there may not be sufficient
volume to extract ten doses from a
single vial.
•
Official Each dose must contain 0.2 mL of
vaccine.
• Discard syringe and needle after
administration to a single patient.
0.2 mL diluted vaccine
• Use a new, sterile needle and syringe
to draw up each new dose.
Information
• If the amount of vaccine remaining
in the vial cannot provide a full dose
of 0.2 mL, discard the vial and any
excess volume.
• Discard any unused vaccine within
12 hours after dilution.
Any unused medicine or waste material should be disposed of in accordance with local
Act
requirements.
1982
7. MEDICINE SCHEDULE
Prescription Medicine.
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 25 of 26
8. SPONSOR
Pfizer New Zealand Limited
P O Box 3998
Auckland, New Zealand
Toll Free Number: 0800 736 363
Released
9. DATE OF FIRST APPROVAL
Date of publication in the New Zealand Gazette of consent to distribute this medicine:
16 December 2021
under
10. DATE OF REVISION OF THE TEXT
22 December 2021
COMIRNATY® is a registered trademark of BioNTech SE. Used under license.
the
Official
Summary of Updates
Section
Update
6.3
Amend shelf life from 6 months to 9 months
6.6
Amend shelf life from 6 months to 9 months
Information
Act 1982
Version: pfdcocii21221
Supersedes: pfdcocii11221
Page 26 of 26
NEW ZEALAND DATA SHEET
1. PRODUCT NAME
COMIRNATY® (orange cap, must dilute), new formulation, 0.1 mg/mL concentrate for
suspension for injection, children 5 to 11 years of age (10 micrograms/0.2 mL dose)
Released
2. QUALITATIVE AND QUANTITATIVE COMPOSITION
This is a multidose vial and
must be diluted before use.
One vial (1.3 mL) contains 10 doses of 0.2 mL after dilution, see Section 4.2 Dose and method
of administration and Section 6.6 Special precautions for disposal and other handling.
under
1 dose (0.2 mL) contains 10 micrograms of tozinameran, a COVID-19 mRNA Vaccine
(embedded in lipid nanoparticles).
Tozinameran is a single-stranded, 5’-capped messenger RNA (mRNA) produced using a cell-
free
in vitro transcription from the corresponding DNA templates, encoding the viral spike (S)
the
protein of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
Official
For the full list of excipients, see Section 6.1 List of excipients.
3. PHARMACEUTICAL FORM
Concentrate for suspension for injection (sterile concentrate).
Information
COMIRNATY is a white to off-white frozen suspension.
4. CLINICAL PARTICULARS
4.1 Therapeutic indications
COMIRNATY (orange cap, must dilute) has provisional consent (see section 5.1) for the
indication below:
Act
Active immunisation to prevent coronavirus disease 2019 (COVID-19) caused by SARS-CoV-
2, in children aged 5 to 11 years.
1982
The use of this vaccine should be in accordance with official recommendations.
4.2 Dose and method of administration
Dose
Children 5 to 11 years of age (i.e. 5 to less than 12 years of age)
COMIRNATY (orange cap, must dilute) is administered intramuscularly as a primary course
of 2 doses (0.2 mL each) at least 21 days apart.
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 1 of 27
The interchangeability of COMIRNATY with other COVID-19 vaccines to complete the
primary vaccination course has not been established. Individuals who have received 1 dose of
COMIRNATY should receive a second dose of COMIRNATY to complete the primary
vaccination course.
COMIRNATY (orange cap, must dilute) should be used only for children 5 to 11 years of age.
Released
Elderly population
Refer to the Data Sheet for COMIRNATY (grey cap, do not dilute), new formulation, 0.1
mg/mL suspension for injection, 12 years of age and older (30 micrograms/dose).
Method of administration
COMIRNATY should be administered intramuscularly, after dilution. The preferred site of
administration is the deltoid muscle of the upper arm.
under
Do not inject COMIRNATY intravascularly, subcutaneously or intradermally.
COMIRNATY should not be mixed in the same syringe with any other vaccines or medicinal
products.
the
For precautions to be taken before administering COMIRNATY, see Section 4.4 Special
warnings and precautions for use.
Official
COMIRNATY (orange cap, must dilute)
Vials have an orange cap and after
dilution contain ten doses of 0.2 mL of vaccine. In order
to extract ten doses from a single vial, low dead-volume syringes and/or needles should be
used. The low dead-volume syringe and needle combination should have a dead volume of no
more than 35 microlitres. If standard syringes and needles are used, there may not be sufficient
Information
volume to extract a tenth dose from a single vial. Irrespective of the type of syringe and needle:
• Each dose must contain 0.2 mL of vaccine.
• If the amount of vaccine remaining in the vial cannot provide a full dose of 0.2 mL, discard
the vial and any excess volume.
• Do not pool excess vaccine from multiple vials.
For instructions on thawing, handling, dilution and dose preparation of COMIRNATY (orange
Act
cap, must dilute) see Section 6.6 Special precautions for disposal and other handling.
4.3 Contraindications
1982
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of
excipients.
4.4 Special warnings and precautions for use
Traceability
In order to improve the traceability of biological medicinal products, the name and the batch
number of the administered product should be clearly recorded.
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 2 of 27
General recommendations
Hypersensitivity and anaphylaxis
Events of anaphylaxis have been reported. Appropriate medical treatment and supervision
should always be readily available in case of an anaphylactic reaction following the
administration of COMIRNATY.
Released
The individual should be kept under close observation for at least 15 minutes following
vaccination. A second dose of COMIRNATY should not be given to those who have
experienced anaphylaxis to the first dose of COMIRNATY.
Myocarditis and pericarditis
Very rare cases of myocarditis and pericarditis have been observed following vaccination with
COMIRNATY. These cases have primarily occurred within 14 days following vaccination,
more often after the second vaccination, and more often in younger men. Available data
under
suggest that the course of myocarditis and pericarditis following vaccination is not different
from myocarditis or pericarditis in general.
Healthcare professionals should be alert to the signs and symptoms of myocarditis and
pericarditis. Vaccinees should be instructed to seek immediate medical attention if they
the
develop symptoms indicative of myocarditis or pericarditis such as (acute and persisting) chest
pain, shortness of breath, or palpitations following vaccination. Healthcare professionals
Official
should consult guidance and/or specialists to diagnose and treat this condition.
Stress-related responses
Some individuals may have stress-related responses associated with the process of vaccination
itself. Stress-related responses are temporary and resolve on their own. They may include
dizziness, fainting, palpitations, increases in heart rate, alterations in blood pressure, feeling
Information
short of breath, tingling sensations, sweating and/or anxiety. Individuals should be advised to
bring symptoms to the attention of the vaccination provider for evaluation and precautions
should be in place to avoid injury from fainting.
Concurrent illness
Vaccination should be postponed in individuals suffering from acute severe febrile illness or
acute infection. The presence of a minor infection and/or low grade fever should not delay
vaccination.
Act
Thrombocytopenia and coagulation disorders
As with other intramuscular injections, COMIRNATY should be given with caution in
1982
individuals receiving anticoagulant ther apy or those with thrombocytopenia or any coagulation
disorder (such as haemophilia) bec ause bleeding or bruising may occur following an
intramuscular administration in these individuals.
Immunocompromised individuals
The efficacy, safety and immunogenicity of COMIRNATY has not been assessed in
immunocompromised individuals, including those receiving immunosuppressant therapy. The
efficacy of COMIRNATY may be lower in immunosuppressed individuals.
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 3 of 27
Duration of protection
The duration of protection afforded by COMIRNATY is unknown as it is still being determined
by ongoing clinical trials.
Limitations of vaccine effectiveness
As with any vaccine, vaccination with COMIRNATY may not protect all vaccine recipients.
Released
Individuals may not be fully protected until 7 days after their second dose of COMIRNATY.
Use in the elderly
Clinical studies of COMIRNATY include participants 65 years of age and older and their data
contributes to the overall assessment of safety and efficacy. See Section 5.1 Pharmacodynamic
properties, Clinical trials, Efficacy against COVID-19.
Paediatric use under
The safety and efficacy of COMIRNATY in children aged less than 5 years of age have not
yet been established.
Effects on laboratory tests the
No data available.
Official
4.5 Interactions with other medicines and other forms of interactions
No interaction studies have been performed.
Concomitant administration of COMIRNATY with other vaccines has not been studied.
Information
4.6 Fertility, pregnancy and lactation
Fertility
In a combined fertility and developmental toxicity study, female rats were intramuscularly
administered COMIRNATY prior to mating and during gestation (4 full human doses of
30 micrograms each, spanning between pre-mating day 21 and gestation day 20). SARS-CoV-
2 neutralising antibodies were present in maternal animals from prior to mating to the end of
the study on postnatal day 21 as well as in fetuses and offspring. There were no vaccine related
Act
effects on female fertility and pregnancy rate.
Pregnancy
1982
There is limited experience with use of COMIRNATY in pregnant women. Animal studies do
not indicate direct or indirect harmful effects with respect to pregnancy, embryo/fetal
development, parturition or post-natal development (see Section 4.6 Fertility, pregnancy and
lactation, Fertility). Administration of COMIRNATY in pregnancy should only be considered
when the potential benefits outweigh any potential risks for the mother and fetus.
Lactation
It is unknown whether tozinameran is excreted in human milk. A combined fertility and
developmental toxicity study in rats did not show harmful effects on offspring development
before weaning (see Section 4.6 Fertility, pregnancy and lactation, Fertility).
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 4 of 27
4.7 Effects on ability to drive and use machines
COMIRNATY has no, or negligible, influence on the ability to drive and use machines.
However, some of the effects mentioned under Section 4.8 Undesirable effects may
temporarily affect the ability to drive or use machines.
4.8 Undesirable effects
Released
Summary of safety profile
The safety of COMIRNATY was evaluated in participants 5 years of age and older in 3 clinical
studies that included 24,675 participants (comprised of 22,026 participants 16 years of age and
older, 1,131 adolescents 12 to 15 years of age and 1,518 children 5 to 11 years of age) that have
received at least one dose of COMIRNATY.
under
Additionally, 306 existing Phase 3 participants at 18 to 55 years of age received a booster dose
of COMIRNATY approximately 6 months after the second dose in the non-placebo-controlled
booster dose portion of Study C4591001. The overall safety profile for the booster dose was
similar to that seen after 2 doses.
the
In Study C4591031, a placebo-controlled booster study, 5,081 participants 16 years of age and
older were recruited from Study C4591001 to receive a booster dose of COMIRNATY at least
6 months after the second dose. The overall safety profile for the booster dose was similar to
Official
that seen after 2 doses.
Participants 16 years of age and older – after 2 doses
In Study C4591001, a total of 22,026 participants 16 years of age or older received at least 1
dose of COMIRNATY 30 micrograms and a total of 22,021 participants 16 years of age or
Information
older received placebo (including 138 and 145 adolescents 16 and 17 years of age in the
COMIRNATY and placebo groups, respectively). A total of 20,519 participants 16 years of
age or older received 2 doses of COMIRNATY.
At the time of the analysis of Study C4591001 with a data cut-off of 13 March 2021 for the
placebo-controlled blinded follow-up period up to the participants’ unblinding dates, a total of
25,651 (58.2%) participants (13,031 COMIRNATY and 12,620 placebo) 16 years of age and
older were followed up for ≥4 months after the second dose. This included a total of 15,111
(7,704 COMIRNATY and 7,407 placebo) participants 16 to 55 years of age and a total of
Act
10,540 (5,327 COMIRNATY and 5,213 placebo) participants 56 years and older.
The most frequent adverse reactions in participants 16 years of age and older that received 2
1982
doses were injection site pain (>80%), fatigue (>60%), headache (>50%), myalgia (>40%),
chills (>30%), arthralgia (>20%), pyrexia and injection site swelling (>10%) and were usually
mild or moderate in intensity and resolved within a few days after vaccination. A slightly lower
frequency of reactogenicity events was associated with greater age.
The safety profile in 545 subjects receiving COMIRNATY, that were seropositive for SARS-
CoV-2 at baseline, was similar to that seen in the general population.
Study C4591001 also included 200 participants with confirmed stable human
immunodeficiency virus (HIV) infection. The safety profile of the participants receiving
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 5 of 27
COMIRNATY (n=100) in the individuals with stable HIV infection was similar to that seen in
the general population.
Adolescents 12 to 15 years of age – after 2 doses
In an analysis of Study C4591001, 2,260 adolescents (1,131 COMIRNATY 30 micrograms;
1,129 placebo) were 12 to 15 years of age. Of these, 1,308 adolescents (660 COMIRNATY
and 648 placebo) have been followed for at least 2 months after the second dose of
Released
COMIRNATY. The safety evaluation in Study C4591001 is ongoing.
The most frequent adverse reactions in adolescents 12 to 15 years of age that received 2 doses
were injection site pain (>90%), fatigue and headache (>70%), myalgia and chills (>40%),
arthralgia and pyrexia (>20%).
Children 5 to 11 years of age – after 2 doses
under
In an analysis of Study C4591007 Phase 2/3, 2,268 children (1,518 COMIRNATY
10 micrograms; 750 placebo) were 5 to 11 years of age. Of these, 2,158 (95.1%)
(1,444 COMIRNATY 10 micrograms and 714 placebo) children have been followed for at
least 2 months after the second dose. The safety evaluation in Study C4591007 is ongoing.
the
The most frequent adverse reactions in children 5 to 11 years of age that received 2 doses
included injection site pain (>80%), fatigue (>50%), headache (>30%), injection site redness
and swelling (>20%), myalgia and chills (>10%).
Official
Participants 16 years of age and older – after booster dose
A subset from Study C4591001 Phase 2/3 participants of 306 adults 18 to 55 years of age who
completed the original COMIRNATY 2-dose course, received a booster dose of
COMIRNATY approximately 6 months (range of 4.8 to 8.0 months) after receiving Dose 2.
Information
The most frequent adverse reactions in participants 18 to 55 years of age were injection site
pain (>80%), fatigue (>60%), headache (>40%), myalgia (>30%), chills and arthralgia (>20%).
In Study C4591031, a placebo-controlled booster study, participants 16 years of age and older
recruited from Study C4591001 received a booster dose of COMIRNATY (5,081 participants),
or placebo (5,044 participants) at least 6 months after the second dose of COMIRNATY.
Overall, participants who received a booster dose, had a median follow-up time of 2.5 months
after the booster dose to the cut-off date (5 October 2021).
Act
Tabulated list of adverse reactions from clinical studies and post-authorisation
experience
1982
Adverse reactions observed during clinical studies are listed below according to the following
frequency categories:
Very common (≥ 1/10),
Common (≥ 1/100 to < 1/10),
Uncommon (≥ 1/1,000 to < 1/100),
Rare (≥ 1/10,000 to < 1/1,000),
Very rare (< 1/10,000),
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 6 of 27
Not known (cannot be estimated from the available data).
Table 1: Adverse reactions from COMIRNATY clinical trials: Individuals 12 years of age
and older
Not known
Rare
Very
Common
Uncommon
(cannot be
System Organ
(≥ 1/10,000
common
(≥ 1/100 to
(≥ 1/1,000 to
estimated from
Released
Class
(≥
to
1/10)
< 1/10)
< 1/100)
the available
< 1/1,000)
data)
Blood and
Lymphadenopathya
lymphatic
system disorders
Metabolism and
Decreased appetite
nutrition
disorders
under
Psychiatric
Insomnia
disorders
Nervous system
Headache
Lethargy
Acute
disorders
peripheral
facial
the
paralysisb
Gastrointestinal
Nausea;
disorders
Official
Skin and
Hyperhidrosis;
subcutaneous
Night sweats
tissue disorders
Musculoskeletal
Arthralgia;
and connective
Myalgia
tissue disorders
Information
General
Injection
Injection
Asthenia; Malaise;
Facial swellingd
disorders and
site pain;
site redness
administration
Fatigue;
site conditions
Chills;
Pyrexiac;
Injection
site
swelling
a A higher frequency of lymphadenopathy (2.8% vs 0.4%) was observed in participants receiving a booster dose
Act
in Study C4591031 compared to participants receiving 2 doses.
b Through the clinical trial safety follow-up period to 14 November 2020, acute peripheral facial paralysis (or
palsy) was reported by four participants in the COMIRNATY group. Onset was Day 37 after Dose 1 (participant
did not receive Dose 2) and Days 3, 9, and 48 after Dose 2. No cases of acute peripheral facial paralysis (or palsy)
1982
were reported in the placebo group.
c A higher frequency of pyrexia was observed after the second dose compared to the first dose. The preferred term
pyrexia is a cluster term covering also body temperature increased..
d Facial swelling in vaccine recipients with a history of injection of dermatological fillers
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 7 of 27
Table 2.
Adverse Reactions from COMIRNATY clinical trial: Individuals 5 to 11
Years of Age (06 September 2021 Data Cut-off Date)
Rare
Common Uncommon
Not known
Very
≥1/100 to ≥1/1,000 to ≥1/10,000
Very Rare
(cannot be
System Organ
Common
to
<1/10
<1/100
<1/10,000
estimated
Class
≥1/10
<1/1,000
(≥1% to
(≥0.1% to
(<0.01%)
from the
(≥10%)
(≥0.01% to
<10%)
<1%)
available data)
Released
<0.1%)
Blood and
Lymphaden
lymphatic system
opathy
disorders
Immune system
Urticariaa,b;
Anaphylaxisa
disorders
Pruritusa,b;
Rasha,b
Metabolism and
Decreased
nutrition disorders
appetite
under
Nervous system
Headache
disorders
Gastrointestinal
Diarrhoeaa; Nausea
disorders
Vomitinga
Musculoskeletal
Myalgia
Arthralgia
Pain in
the
and connective
extremity
tissue disorders
(arm)a
General disorders
Injection
Pyrexia
Malaise
Official
and administration site pain;
site conditions
Fatigue;
Chills;
Injection
site
swelling;
Information
Injection
site redness
a. These adverse reactions were identified in the post-authorisation period. The following events
were not reported in participants 5 to 11 Years of Age in Study C4591007 but were reported in
individuals ≥16 years of age in Study C4591001: angioedema, lethargy, asthenia, hyperhidrosis,
and night sweats.
b. The following events are categorised as hypersensitivity reactions: urticaria, pruritus, and rash
Post-marketing experience
Act
Although the events listed in Table 3 were not observed in the clinical trials, they are considered
adverse drug reactions for COMIRNATY as they were reported in the post-marketing
experience. As these reactions were derived from spontaneous reports, the frequencies could
1982
not be determined and are thus considered as not known.
Table 3: Adverse reactions from COMIRNATY post marketing experience
System Organ Class
Adverse Drug Reaction
Immune system disorders
Anaphylaxis
Hypersensitivity reactions (e.g. rash, pruritis, urticaria, angioedema)
Cardiac disorders
Myocarditis
Pericarditis
Gastrointestinal disorders
Diarrhoea
Vomiting
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 8 of 27
System Organ Class
Adverse Drug Reaction
Musculoskeletal and connective
Pain in extremity (arm)a
tissue disorders
General disorders and
Extensive swelling of vaccinated limb
administration site conditions
a A higher frequency of pain in extremity (1.1% vs. 0.8%) was observed in participants receiving a booster dose in
Study C4591031 compared to participants receiving 2 doses.
Released
Reporting suspected adverse effects
Reporting suspected adverse reactions after authorisation of the medicine is important. It allows
continued monitoring of the benefit/risk balance of the medicine. Healthcare professionals are
asked to report any suspected adverse reactions at
https://nzphvc.otago.ac.nz/reporting/.
4.9 Overdose
under
Overdose data is available from 52 study participants included in the clinical trial that due to
an error in dilution received 58 micrograms of COMIRNATY. The COMIRNATY recipients
did not report an increase in reactogenicity or adverse reactions.
In the event of overdose, monitoring of vital functions and possible symptomatic treatment is
the
recommended.
Official
For advice on the management of overdose please contact the National Poisons Centre on 0800
POISON (0800 764766).
5. PHARMACOLOGICAL PROPERTIES
Information
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: vaccines, other viral vaccines, ATC code: J07BX03.
Mechanism of action
The nucleoside-modified messenger RNA in COMIRNATY is formulated in lipid
nanoparticles, which enable delivery of the non-replicating RNA into host cells to direct
transient expression of the SARS-CoV-2 spike (S) antigen. The mRNA codes for membrane-
anchored, full-length S with two point mutations within the central helix. Mutation of these
Act
two amino acids to proline locks S in an antigenically preferred prefusion conformation.
COMIRNATY elicits both neutralising antibody and cellular immune responses to the antigen,
which may contribute to protection against COVID-19.
1982
Clinical efficacy and safety
Efficacy
Study C4591001 is a multicentre, multinational, Phase 1/2/3 randomised, placebo-controlled,
observer-blind dose-finding, vaccine candidate selection and efficacy study in participants 12
years of age and older. Randomisation was stratified by age: 12 to 15 years of age, 16 to 55
years of age, or 56 years of age and older, with a minimum of 40% of participants in the ≥56-
year stratum. The study excluded participants who were immunocompromised and those who
had previous clinical or microbiological diagnosis of COVID-19. Participants with pre-existing
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 9 of 27
stable disease, defined as disease not requiring significant change in therapy or hospitalisation
for worsening disease during the 6 weeks before enrolment, were included as were participants
with known stable infection with HIV, hepatitis C virus (HCV) or hepatitis B virus (HBV).
Efficacy in participants 16 years of age and older – after 2 doses
In the Phase 2/3 portion of Study C4591001, based on data accrued through
14 November 2020, approximately 44,000 participants were randomised equally and were to
Released
receive 2 doses of COMIRNATY or placebo. The efficacy analyses included participants that
received their second vaccination within 19 to 42 days after their first vaccination. The majority
(93.1%) of vaccine recipients received the second dose 19 days to 23 days after Dose 1.
Participants are planned to be followed for up to 24 months after Dose 2, for assessments of
safety and efficacy against COVID-19. In the clinical study, participants were required to
observe a minimum interval of 14 days before and after administration of an influenza vaccine
in order to receive either placebo or COMIRNATY. In the clinical study, participants were
under
required to observe a minimum interval of 60 days before or after receipt of blood/plasma
products or immunoglobulins through to conclusion of the study in order to receive either
placebo or COMIRNATY.
The population for the analysis of the primary efficacy endpoint included 36,621 participants
the
12 years of age and older (18,242 in the COMIRNATY group and 18,379 in the placebo group)
who did not have evidence of prior infection with SARS-CoV-2 through 7 days after the second
dose. In addition, 134 participants were between the ages of 16 to 17 years of age (66 in the
Official
COMIRNATY group and 68 in the placebo group) and 1616 participants 75 years of age and
older (804 in the COMIRNATY group and 812 in the placebo group).
At the time of the primary efficacy analysis, participants had been followed for symptomatic
COVID-19 for in total 2,214 person-years for the COMIRNATY group and in total 2,222
person-years for the placebo group.
Information
There were no meaningful clinical differences in overall vaccine efficacy in participants who
were at risk of severe COVID-19 including those with 1 or more comorbidities that increase
the risk of severe COVID-19 (e.g. asthma, body mass index (BMI) ≥30 kg/m2, chronic
pulmonary disease, diabetes mellitus, hypertension).
COMIRNATY efficacy information is presented in Table 4.
Table 4: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2, by age
Act
subgroup – participants without evidence of infection prior to 7 days after Dose 2 –
evaluable efficacy (7 days) population
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
1982
of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Na = 18,198 Cases
Na = 18,325 Cases
Vaccine efficacy
Subgroup
n1b
n1b
%
Surveillance timec
Surveillance timec
(95% CI)f
(n2d)
(n2d)
All participantse
8
162
95.0
2.214 (17,411)
2.222 (17,511)
(90.0, 97.9)
16 to 64 years
7
143
95.1
1.706 (13,549)
1.710 (13,618)
(89.6, 98.1)
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 10 of 27
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Na = 18,198 Cases
Na = 18,325 Cases
Vaccine efficacy
Subgroup
n1b
n1b
%
Surveillance timec
Surveillance timec
(95% CI)f
(n2d)
(n2d)
Released
65 years and older
1
19
94.7
0.508 (3848)
0.511 (3880)
(66.7, 99.9)
65 to 74 years
1
14
92.9
0.406 (3074)
0.406 (3095)
(53.1, 99.8)
75 years and older
0
5
100.0
0.102 (774)
0.106 (785)
(-13.1, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 [*Case definition: (at least 1 of) fever, new or increased cough,
under
new or increased shortness of breath, chills, new or increased muscle pain, new loss of taste or smell, sore
throat, diarrhoea or vomiting.]
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the last dose) of
past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not
detected by nucleic acid amplification tests (NAAT) [nasal swab] at Visits 1 and 2), and had negative NAAT
the
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
Official
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of
the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. No confirmed cases were identified in adolescents 12 to 15 years of age.
f. Two-sided confidence interval (CI) for vaccine efficacy (VE) is derived based on the Clopper and Pearson
method adjusted to the surveillance time. CI not adjusted for multiplicity.
Information
In the second primary analysis, efficacy of COMIRNATY in preventing first COVID-19
occurrence from 7 days after Dose 2 compared to placebo was 94.6% (95% credible interval
of 89.9% to 97.3%) in participants 16 years of age and older with or without evidence of prior
infection with SARS-CoV-2.
Additionally, subgroup analyses of the primary efficacy endpoint showed similar efficacy point
estimates across genders, ethnic groups, and participants with medical comorbidities associated
with high risk of severe COVID-19.
Act
Updated efficacy analyses were performed with additional confirmed COVID-19 cases accrued
during blinded placebo-controlled follow-up through 13 March 2021, representing up to
6 months of follow-up after Dose 2 for participants in the efficacy population.
1982
The updated vaccine efficacy information is presented in Table 5.
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 11 of 27
Table 5: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2, by age
subgroup – participants without evidence of infection prior to 7 days after Dose 2 –
evaluable efficacy (7 days) population during the placebo-controlled follow-up period
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Na=20,998
Na=21,096
Released
Cases
Cases
n1b
n1b
Surveillance Timec
Surveillance Timec
Vaccine efficacy %
Subgroup
(n2d)
(n2d)
(95% CIe)
All participantsf
77
850
91.3
6.247 (20,712)
6.003 (20,713)
(89.0, 93.2)
16 to 64 years
70
710
90.6
4.859 (15,519)
4.654 (15,515)
(87.9, 92.7)
under
65 years and older
7
124
94.5
1.233 (4192)
1.202 (4226)
(88.3, 97.8)
65 to 74 years
6
98
94.1
0.994 (3350)
0.966 (3379)
(86.6, 97.9)
75 years and older
1
26
96.2
the
0.239 (842)
0.237 (847)
(76.9, 99.9)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
Official
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative
at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
Information
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Two-sided confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method
adjusted to the surveillance time.
f. Included confirmed cases in participants 12 to 15 years of age: 0 in the COMIRNATY group (both without
and with or without evidence of prior SARS-CoV-2 infection); 16 and 18 in the placebo group (without and
with or without evidence of prior SARS-CoV-2 infection, respectively).
Act
Efficacy against severe COVID-19 in participants 12 years of age or older – after 2 doses
As of 13 March 2021, vaccine efficacy against severe COVID-19 is presented only for
participants with or without prior SARS-CoV-2 infection (Table 6) as the COVID-19 case
1982
counts in participants without prior SARS-CoV-2 infection were the same as those in
participants with or without prior SARS-CoV-2 infection in both the COMIRNATY and
placebo groups.
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 12 of 27
Table 6. Vaccine Efficacy – First Severe COVID-19 Occurrence in Participants With
or Without* Prior SARS-CoV-2 Infection Based on Food and Drug Administration
(FDA)† Definition After Dose 1 or From 7 Days After Dose 2 in the Placebo-Controlled
Follow-up
COMIRNATY
Placebo
Cases
Cases
Released
n1a
n1a
Vaccine Efficacy %
Surveillance Time (n2b) Surveillance Time (n2b)
(95% CIc)
1
30
96.7
After Dose 1d
8.439e (22,505)
8.288e (22,435)
(80.3, 99.9)
1
21
95.3
7 days after Dose 2f
6.522g (21,649)
6.404g (21,730)
(70.9, 99.9)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at
least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or increased
shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat; diarrhoea;
under
vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative
at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
† Severe illness from COVID-19 as defined by FDA is confirmed COVID-19 and presence of at least 1 of the
the
following:
• Clinical signs at rest indicative of severe systemic illness (respiratory rate ≥30 breaths per minute, heart
rate ≥125 beats per minute, saturation of oxygen ≤93% on room air at sea level, or ratio of arterial
oxygen partial pressure to fractional inspired oxygen <300 mm Hg);
Official
• Respiratory failure [defined as needing high-flow oxygen, noninvasive ventilation, mechanical
ventilation or extracorporeal membrane oxygenation (ECMO)];
• Evidence of shock (systolic blood pressure <90 mm Hg, diastolic blood pressure <60 mm Hg, or
requiring vasopressors);
• Significant acute renal, hepatic, or neurologic dysfunction;
• Admission to an Intensive Care Unit;
Information
• Death.
a. n1 = Number of participants meeting the endpoint definition.
b. n2 = Number of participants at risk for the endpoint.
c. Two-side confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method
adjusted to the surveillance time.
d. Efficacy assessed based on the Dose 1 all available efficacy (modified intention-to-treat) population that
included all randomised participants who received at least 1 dose of study intervention.
e. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from Dose 1 to the end of the surveillance
period.
f. Efficacy assessed based on the evaluable efficacy (7 Days) population that included all eligible randomised
Act
participants who receive all dose(s) of study intervention as randomised within the predefined window, have
no other important protocol deviations as determined by the clinician
g. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
1982
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
Efficacy and immunogenicity in adolescents 12 to 15 years of age – after 2 doses
An analysis of Study C4591001 has been performed in adolescents 12 to 15 years of age up to
a data cutoff date of 13 March 2021.
The vaccine efficacy information in adolescents 12 to 15 years of age is presented in Table 7.
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 13 of 27
Table 7: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2 –
participants without evidence of infection and with or without evidence of infection prior
to 7 days after Dose 2 – adolescents 12 to 15 years of age evaluable efficacy (7 days)
population
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
without evidence of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Released
Na = 1005
Na = 978
Cases
Cases
n1b
n1b
Vaccine efficacy
Surveillance timec (n2d)
Surveillance timec (n2d)
% (95% CIe)
Adolescents
0
16
12 to 15 years
0.154 (1001)
0.147 (972)
100.0 (75.3, 100.0)
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
with or without* evidence of prior SARS-CoV-2 infection
under
COMIRNATY
Placebo
Na = 1119
Na = 1110
Cases
Cases
n1b
n1b
Vaccine efficacy
Surveillance timec (n2d)
Surveillance timec (n2d)
% (95% CIe)
the
Adolescents
0
18
12 to 15 years
0.170 (1109)
0.163 (1094)
100.0 (78.1, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at
Official
least 1 symptom consistent with COVID-19 [*Case definition: (at least 1 of) fever, new or increased cough, new
or increased shortness of breath, chills, new or increased muscle pain, new loss of taste or smell, sore throat,
diarrhoea or vomiting).
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the last dose) of
past SARS-CoV-2 infection (i.e, N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not
detected by nucleic acid amplification tests (NAAT) [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
Information
a. N = number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all subjects within each group at
risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of subjects at risk for the endpoint.
e. Confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method adjusted
for surveillance time. CI not adjusted for multiplicity.
In Study C4591001 an analysis of SARS-CoV-2 neutralising titres in a randomly selected
Act
subset of participants was performed to demonstrate non-inferior immune responses (within
1.5-fold) comparing adolescents 12 to 15 years of age to participants 16 to 25 years of age who
had no serological or virological evidence of past SARS-CoV-2 infection. The immune
1982
response to COMIRNATY in adolescents 12 to 15 years of age (n = 190) was non-inferior to
the immune response in participants 16 to 25 years of age (n = 170), based on results for SARS-
CoV-2 neutralising titres at 1 month after Dose 2. The geometric mean titres (GMT) ratio of
the adolescents 12 to 15 years of age group to the participants 16 to 25 years of age group was
1.76, with a 2-sided 95% CI of 1.47 to 2.10, meeting the 1.5-fold non-inferiority criterion (the
lower bound of the 2-sided 95% CI for the geometric mean ratio [GMR] >0.67), which
indicates a statistically greater response in the adolescents 12 to 15 years of age than that of
participants 16 to 25 years of age.
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 14 of 27
Efficacy in children 5 to <12 years of age – after 2 doses
A descriptive efficacy analysis of Study C4591007 has been performed in 1,968 children 5 to
11 years of age without evidence of infection prior to 7 days after Dose 2. This analysis
evaluated confirmed symptomatic COVID-19 cases accrued up to a data cut-off date of 8
October 2021.
The descriptive vaccine efficacy results in children 5 to 11 years of age without evidence of
Released
prior SARS-CoV-2 infection are presented in Table 8. None of the cases accrued met criteria
for severe COVID-19 or multisystem inflammatory syndrome in children (MIS-C). No cases
of COVID-19 were observed in either the vaccine group or the placebo group in participants
with evidence of prior SARS-CoV-2 infection.
Table 8: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Dose 2:
Without Evidence of Infection Prior to 7 Days After Dose 2 – Phase 2/3 – Children 5 To
under
11 Years of Age Evaluable Efficacy Population
First COVID-19 occurrence from 7 days after Dose 2 in children 5 to 11 years of age
without evidence of prior SARS-CoV-2 infection*
COMIRNATY±
the
10 micrograms/dose
Placebo
Na=1305
Na=663
Cases
Cases
n1b
n1b
Official
Surveillance Timec
Surveillance Timec
Vaccine Efficacy %
(n2d)
(n2d)
(95% CI)
Children 5 to
3
16
90.7
11 years of age
0.322 (1273)
0.159 (637)
(67.7, 98.3)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
Information
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
*
Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum]
negative at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had
negative NAAT (nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the
analysis.
± Pfizer-BioNTech COVID-19 Vaccine (10 micrograms modRNA).
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each
Act
group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the
end of the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
1982
Immunogenicity in children 5 to 11 years of age – after 2 doses
Study C4591007 is a Phase 1/2/3 study comprised of an open-label vaccine dose-finding
portion (Phase 1) and a multicentre, multinational, randomised, saline placebo-controlled,
observer-blind efficacy portion (Phase 2/3) that has enrolled participants 5 to 11 years of age.
In C4591007, an analysis of SARS-CoV-2 50% neutralising titres (NT50) 1 month after Dose
2 in a randomly selected subset of participants demonstrated effectiveness by immunobridging
of immune responses comparing children 5 to 11 years of age in the Phase 2/3 part of Study
C4591007 to participants 16 to 25 years of age in the Phase 2/3 part of Study C4591001 who
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 15 of 27
had no serological or virological evidence of past SARS-CoV-2 infection up to 1 month after
Dose 2, meeting the prespecified immunobridging criteria for both the geometric mean ratio
(GMR) and the seroresponse difference with seroresponse defined as achieving at least 4-fold
rise in SARS-CoV-2 NT50 from baseline (before Dose 1).
The ratio of the SARS-CoV-2 NT50 in children 5 to 11 years of age to that of young adults 16
to 25 years of age was 1.04 (2-sided 95% CI: 0.93, 1.18), as presented in Table 9.
Released
Table 9: Summary of geometric mean ratio for 50% neutralising titre – Comparison of
children 5 to 11 years of age (Study C4591007) to participants 16 to 25 years of age (Study
C4591001) – participants without* evidence of infection up to 1 month after Dose 2 –
evaluable immunogenicity population
COMIRNATY
5 to 11 years/
10 microgram/dose 30 microgram/dose
16 to 25 years
5 to 11 years
16 to 25 years
under
na=264
na=253
Met
Time
GMTc
GMTc
GMRd
immunobridging
Assay
pointb
(95% CIc)
(95% CIc)
(95% CId)
objectivee
(Y/N)
the
SARS-CoV-2
neutralisation 1 month
1197.6
1146.5
1.04
Y
assay - NT50 after
(1106.1, 1296.6)
(1045.5, 1257.2)
(0.93, 1.18)
Official
(titre)f
Dose 2
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre;
LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test; NT50 = 50% neutralising titre;
SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
*Participants who had no serological or virological evidence (up to 1 month post-Dose 2 blood sample collection)
of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and 1 month after Dose
Information
2, SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2, and negative NAAT (nasal swab) at
any unscheduled visit up to 1 month after Dose 2 blood collection) and had no medical history of COVID-
19 were included in the analysis.
a. n = Number of participants with valid and determinate assay results for the specified assay at the given
dose/sampling time point.
b. Protocol-specified timing for blood sample collection.
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
d. GMRs and 2-sided 95% CIs were calculated by exponentiating the mean difference of the logarithms of the
titres (Group 1[5 to 11 years of age] - Group 2 [16 to 25 years of age]) and the corresponding CI (based on
Act
the Student t distribution).
e. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67 and
the point estimate of the GMR is ≥0.8.
f. SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation
1982
Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus
neutralisation is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution
at which 50% of the virus is neutralised.
Among participants without prior evidence of SARS-CoV-2 infection up to 1 month after
Dose 2, 99.2% of children 5 to 11 years of age and 99.2% of participants 16 to 25 years of age
had a seroresponse from before vaccination to 1 month after Dose 2. The difference in
proportions of participants who had seroresponse between the 2 age groups (children – young
adult) was 0.0% (2-sided 95% CI: -2.0%, 2.2%) as presented in Table 10.
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 16 of 27
Table 10: Difference in percentages of participants with seroresponse – participants
without evidence of infection up to 1 month after Dose 2 – immunobridging subset –
Phase 2/3 – comparison of 5 to 11 years of age to Study C4591001 Phase 2/3 16 to 25 years
of age – evaluable immunogenicity population
COMIRNATY
10
30
5 to 11 years/
microgram/dose microgram/dose
Released
16 to 25 years
5 to 11 years
16 to 25 years
Na=264
Na=253
Met
Time
nc (%)
nc (%)
Difference %e immunobridging
Assay
pointb
(95% CId)
(95% CId)
(95% CIf)
objectiveg
(Y/N)
SARS-CoV-2
1 month
neutralisation
262 (99.2)
251 (99.2)
0.0
under
after
Y
assay – NT50
(97.3, 99.9)
(97.2, 99.9)
(-2.0, 2.2)
Dose 2
(titre)h
Abbreviations: LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test;
N-binding = SARS-CoV-2 nucleoprotein–binding; NT50 = 50% neutralising titre 50; SARS-CoV-2 = severe acute
respiratory syndrome coronavirus 2.
Note: Seroresponse is defined as achieving a ≥4
the -fold rise from baseline (before Dose 1). If the baseline measurement
is below the LLOQ, a postvaccination assay result ≥4 × LLOQ is considered a seroresponse.
Note: Participants who had no serological or virological evidence (up to 1 month post-Dose 2 blood sample
collection) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and 1 month after
Official
Dose 2, SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2, and negative NAAT (nasal swab) at
any unscheduled visit up to 1 month after Dose 2 blood collection) and had no medical history of COVID-19 were
included in the analysis.
a. N = number of participants with valid and determinate assay results both before vaccination and at 1 month after
Dose 2. These values are the denominators for the percentage calculations.
b. Protocol-specified timing for blood sample collection.
c. n = Number of participants with seroresponse for the given assay at the given dose/sampling time point.
Information
d. Exact 2-sided CI based on the Clopper and Pearson method.
e. Difference in proportions, expressed as a percentage (Group 1 [5 to 11 years of age] – Group 2 [16 to 25 years
of age]).
f. 2-Sided CI, based on the Miettinen and Nurminen method for the difference in proportions, expressed as a
percentage.
g. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the difference in proportions is greater
than -10.0%.
h. SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation Assay.
The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus neutralisation is
read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution at which 50% of
Act
the virus is neutralised.
Immunogenicity in participants 18 years of age and older – after booster dose
1982
Effectiveness of a booster dose of COMIRNATY was based on an assessment of 50%
neutralising titres (NT50) against SARS-CoV-2 (USA_WA1/2020). In Study C4591001,
analyses of NT50 1 month after the booster dose compared to 1 month after the primary series
in individuals 18 to 55 years of age who had no serological or virological evidence of past
SARS-CoV-2 infection up to 1 month after the booster vaccination demonstrated
noninferiority for both GMR and difference in seroresponse rates. Seroresponse for a
participant was defined as achieving a ≥4-fold rise in NT50 from baseline (before Dose 1),
These analyses are summarised in Table 11.
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 17 of 27
Table 11. SARS-CoV-2 neutralisation assay - NT50 (titre)† (SARS-CoV-2
USA_WA1/2020) – GMT and seroresponse rate comparison of 1 month after booster dose
to 1 month after primary series – participants 18 to 55 years of age without evidence of
infection up to 1 month after booster dose* – booster dose evaluable immunogenicity
population±
1 month after
booster dose/-
Released
1 month after
Met
1 month after
1 month after
primary
noninferiority
booster dose
primary series
series
objective
n
(95% CI)
(95% CI)
(97.5% CI)
(Y/N)
Geometric mean
50% neutralising
2466.0b
750.6
b
3.29c
titre (GMTb)
212a
(2202.6, 2760.8)
(656.2, 858.6)
(2.77, 3.90)
Yd
Seroresponse rate
199f
196f
under
(%) for 50%
99.5%
98.0%
1.5%g
neutralising titre†
200e
(97.2%, 100.0%) (95.0%, 99.5%) (-0.7%, 3.7%
h)
Yi
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre;
LLOQ = lower limit of quantitation; N-binding = SARS-CoV-2 nucleoprotein-binding; NAAT = nucleic acid
amplification test; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute respiratory syndrome
coronavirus 2; Y/N = yes/no.
the
†
SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation
Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus
neutralisation is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum
Official
dilution at which 50% of the virus is neutralised.
*
Participants who had no serological or virological evidence (up to 1 month after receipt of a booster dose
of Comirnaty) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative and
SARS-CoV-2 not detected by NAAT [nasal swab]) and had a negative NAAT (nasal swab) at any
unscheduled visit up to 1 month after the booster dose were included in the analysis.
± All eligible participants who had received 2 doses of Comirnaty as initially randomised, with Dose 2
received within the predefined window (within 19 to 42 days after Dose 1), received a booster dose of
Information
Comirnaty, had at least 1 valid and determinate immunogenicity result after booster dose from a blood
collection within an appropriate window (within 28 to 42 days after the booster dose), and had no other
important protocol deviations as determined by the clinician.
a. n = Number of participants with valid and determinate assay results at both sampling time points within
specified window.
b. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
c. GMRs and 2-sided 97.5% CIs were calculated by exponentiating the mean differences in the logarithms of
the assay and the corresponding CIs (based on the Student t distribution).
d. Noninferiority is declared if the lower bound of the 2-sided 97.5% CI for the GMR is > 0.67 and the point
Act
estimate of the GMR is ≥ 0.80.
e. n = Number of participants with valid and determinate assay results for the specified assay at baseline,
1 month after Dose 2 and 1 month after the booster dose within specified window. These values are the
1982
denominators for the percentage calculations.
f. Number of participants with seroresponse for the given assay at the given dose/sampling time point. Exact
2-sided CI based on the Clopper and Pearson method.
g. Difference in proportions, expressed as a percentage (1 month after booster dose – 1 month after Dose 2).
h. Adjusted Wald 2-sided CI for the difference in proportions, expressed as a percentage.
i. Noninferiority is declared if the lower bound of the 2-sided 97.5% CI for the percentage difference is
> -10%.
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 18 of 27
Relative vaccine efficacy in participants 16 years of age and older – after booster dose
An interim efficacy analysis of Study C4591031, a placebo-controlled booster study, was
performed in approximately 10,000 participants 16 years of age and older who were recruited
from Study C4591001, evaluated confirmed COVID-19 cases accrued from at least 7 days after
Released
booster vaccination up to a data cut-off date of 5 October 2021, which represents a median of
2.5 months post-booster follow-up. Vaccine efficacy of the COMIRNATY booster dose after
the primary series relative to the placebo booster group who only received the primary series
dose was assessed. The relative vaccine efficacy information for participants 16 years of age
and older is presented in Table 12.
Table 12: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Booster
under
Vaccination – Participants 16 Years of Age and Older With or Without
Evidence of Infection Prior to 7 Days After Booster Vaccination – Evaluable
Efficacy Population
First COVID-19 occurrence from 7 days after booster dose in participants with or without
the
evidence of prior SARS-CoV-2 infection
COMIRNATY
Placebo
Na=4993
Na=4952
Cases
Cases
Official
n1b
n1b
Relative Vaccine
Surveillance Timec
Surveillance Timec
Efficacye %
(n2d)
(n2d)
(95% CIf)
First COVID-19
occurrence from 7
days after booster
7
124
94.6
Information
vaccination
0.871 (4934)
0.835 (4863)
(88.5, 97.9)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each
group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after the booster
Act
vaccination to the end of the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Relative vaccine efficacy of the Comirnaty booster group relative to the placebo group (non-booster).
f. Two-sided confidence interval (CI) for relative vaccine efficacy is derived based on the Clopper and 1982
Pearson method adjusted for surveillance time.
This medicine has been given a provisional consent under Section 23 of the Act. This means
that further evidence on this medicine is awaited or that there are specific conditions of use.
Refer to the consent notice published in the New Zealand Gazette for the specific conditions.
5.2 Pharmacokinetic properties
Not applicable.
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 19 of 27
5.3 Preclinical safety data
Genotoxicity/Carcinogenicity
Neither genotoxicity nor carcinogenicity studies were performed. The components of
COMIRNATY (lipids and mRNA) are not expected to have genotoxic potential.
Released
6. PHARMACEUTICAL PARTICULARS
6.1 List of excipients
((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
Distearoylphosphatidylcholine (DSPC)
under
Cholesterol
Trometamol
Trometamol hydrochloride the
Sucrose
Water for injections
Official
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those
mentioned in Section 6.6 Special precautions for disposal and other handling.
Information
6.3 Shelf life
COMIRNATY (orange cap, must dilute)
Unopened vial
Frozen vial
12 months when stored at -90°C to -60°C.
Act
The vaccine will be received frozen at -90°C to -60°C. Frozen vaccine can be stored either at
-90°C to -60°C or 2°C to 8°C upon receipt.
1982
When stored frozen at -90°C to -60°C, 10-vial packs of the vaccine can be thawed at 2°C to 8°C
for 4 hours or individual vials can be thawed at room temperature (up to 30°C) for 30 minutes.
Thawed vial
If the vaccine is received at 2°C to 8°C it should be stored at 2°C to 8°C. Once removed from
frozen storage, the unopened vial may be stored refrigerated at 2°C to 8°C for a single period
of up to 10 weeks within the 12-month shelf life.
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 20 of 27
Upon moving the product to 2°C to 8°C storage, the updated expiry date must be written on
the outer carton and the vaccine should be used or discarded by the updated expiry date. The
original expiry date should be crossed out.
Check that the expiry date on the outer carton has been updated to reflect the refrigerated expiry
date and that the original expiry date has been crossed out.
Released
When stored frozen at -90°C to -60°C, the vaccine can be thawed at either 2°C to 8°C or at
temperatures up to 30°C.
Prior to use, the unopened vials can be stored for up to 12 hours at temperatures between
8ºC to 30ºC.
Thawed vials can be handled in room light conditions.
under
Once thawed COMIRNATY (orange cap, must dilute) should not be re-frozen.
Diluted medicinal product
Chemical and physical in-use stability has been demonstrated for 12 hours at 2ºC to 30°C, after
dilution with sodium chloride 9 mg/mL (0.9%) solution for injection. From a microbiological
the
point of view, unless the method of dilution precludes the risk of microbial contamination, the
product should be used immediately. If not used immediately, in-use storage times and
conditions are the responsibility of the user.
Official
6.4 Special precautions for storage
COMIRNATY (orange cap, must dilute) can be stored in a refrigerator at 2°C to 8°C for a
single period of up to 10 weeks, not exceeding the original expiry date (EXP). The expiry date
Information
for storage at -90°C to -60°C is printed on the vial and outer carton after “EXP”.
Check that the expiry date has been updated to reflect the refrigerated EXP date and that the
original expiry date has been crossed out.
Store in the original package to protect from light. During storage, minimise exposure to room
light, and avoid exposure to direct sunlight and ultraviolet light.
For detailed instructions see Section 6.6 Special precautions for disposal and other handling.
Act
Once thawed, the vaccine cannot be re-frozen.
1982
Thawed vials can be handled in room light conditions.
For storage conditions after thawing and dilution of the medicinal product, see Section 6.3
Shelf life.
For additional advice on storing COMIRNATY, contact Pfizer New Zealand on 0800 736 363.
6.5 Nature and contents of container
COMIRNATY (orange cap, must dilute) 1.3 mL fill volume in 2 mL clear multidose vial
(Type I glass) with a stopper (synthetic bromobutyl rubber) and an orange flip-off plastic cap
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 21 of 27
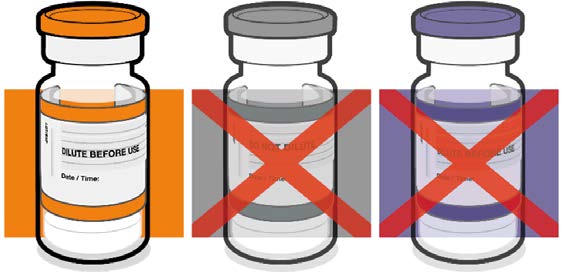
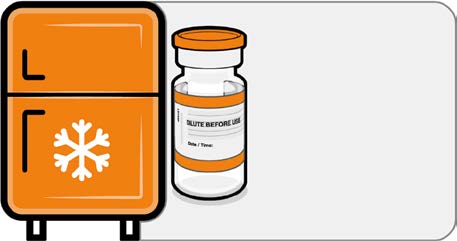


with aluminium seal. Each vial contains 10 doses, see Section 6.6 Special precautions for
disposal and other handling.
Pack size: 10 vials, 195 vials
Not all pack sizes may be marketed.
Released
6.6 Special precautions for disposal and other handling
COMIRNATY (orange cap, must dilute)
The vaccine should be prepared by a healthcare professional using aseptic technique to ensure
the sterility of the prepared diluted suspension.
COMIRNATY (orange cap, must dilute)
under
Dose Verification
• Verify that the vial has an orange
plastic cap.
Orange cap
the
• Only the orange cap vial can be used
for children age 5 to 11 years.
Official
10 micrograms
Information
Handling Prior To Use
• If the multidose vial is stored frozen
it must be thawed prior to use.
Frozen vials should be transferred to
an environment of 2°C to 8°C to
thaw; a 10 vial pack may take
4 hours to thaw. Ensure vials are
Store for up to
completely thawed prior to use.
10 weeks at
•
Act
Unopened vials can be stored for up
2 °C to 8 °C
to 10 weeks at 2°C to 8°C within the
12 month shelf life.
1982
• Alternatively, individual frozen vials
may be thawed for 30 minutes at
temperatures up to 30°C for
immediate use.
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 22 of 27
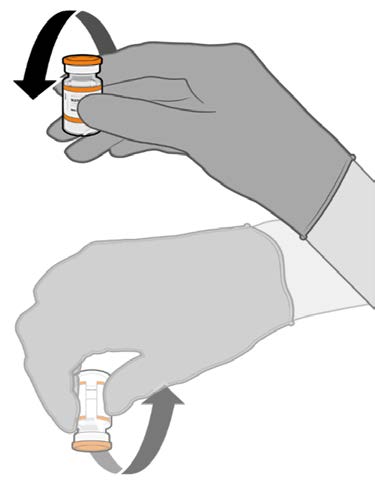
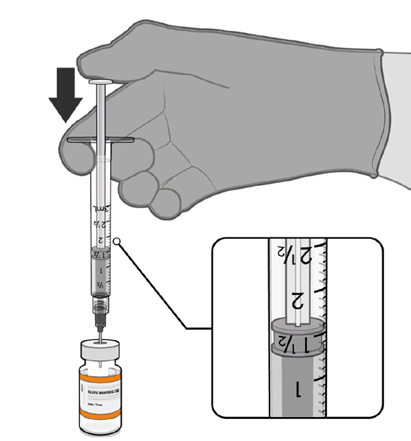 COMIRNATY (orange cap, must dilute)
Mixing Prior To Dilution
COMIRNATY (orange cap, must dilute)
Mixing Prior To Dilution
• Allow the thawed vial to come to
room temperature and gently invert it
10 times prior to dilution. Do not
Released
shake.
• Prior to dilution, the thawed
suspension may contain white to off-
white opaque amorphous particles.
under
the
Official
Dilution
• The thawed vaccine must be diluted
in its original vial with 1.3 mL
sodium chloride 9 mg/mL (0.9%)
solution for injection, using a
Information
21 gauge or narrower needle and
aseptic techniques.
Act 1982
1.3 mL of 0.9% sodium chloride
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 23 of 27
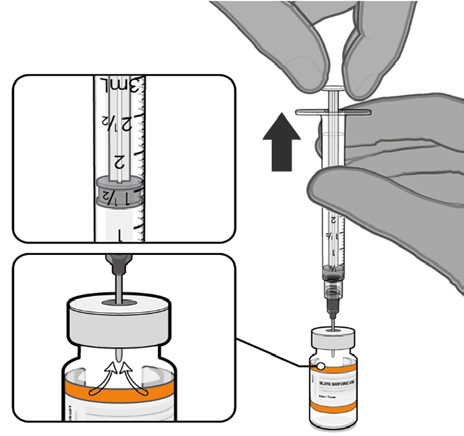
 COMIRNATY (orange cap, must dilute)
Dilution (continued)
COMIRNATY (orange cap, must dilute)
Dilution (continued)
• Equalise vial pressure before
removing the needle from the vial
stopper by withdrawing 1.3 mL air
Released
into the empty diluent syringe.
under
the
Pull back plunger to 1.3 mL to
remove air from vial.
Official
• Gently invert the diluted suspension
10 times. Do not shake.
• The diluted vaccine should present as
a white to off-white suspension with
no particulates visible. Do not use
Information
the diluted vaccine if particulates or
discoloration are present.
Act 1982
Gently × 10
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 24 of 27
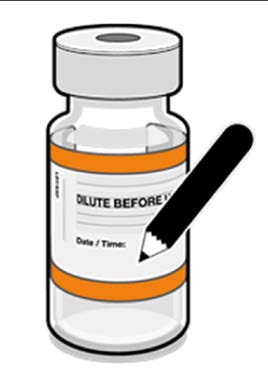 COMIRNATY (orange cap, must dilute)
Dilution (continued)
COMIRNATY (orange cap, must dilute)
Dilution (continued)
• The diluted vials should be marked
with the appropriate date and time.
• After dilution, store at 2°C to 30°C
Released
and use within 12 hours.
• Do not freeze or shake the diluted
dispersion. If refrigerated, allow the
diluted suspension to come to room
temperature prior to use.
under
Record appropriate date and time.
Use within 12 hours after dilution.
the
Official
Information
Act 1982
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 25 of 27
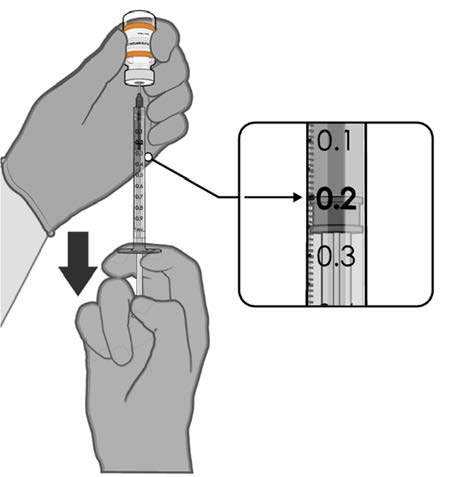 COMIRNATY (orange cap, must dilute)
Preparation of Individual 0.2 mL Doses of COMIRNATY (orange cap, must dilute)
COMIRNATY (orange cap, must dilute)
Preparation of Individual 0.2 mL Doses of COMIRNATY (orange cap, must dilute)
• Using aseptic technique, cleanse the
vial stopper with a single-use
antiseptic swab.
Released
• Withdraw 0.2 mL of COMIRNATY
(orange cap, must dilute).
Low dead-volume syringes and/or
needles should be used in order to
extract 10 doses from a single vial.
The low dead-volume syringe and
needle combination should have a
under
dead volume of no more than
35 microlitres.
If standard syringes and needles are
the
used, there may not be sufficient
volume to extract ten doses from a
single vial.
•
Official Each dose must contain 0.2 mL of
vaccine.
• Discard syringe and needle after
administration to a single patient.
0.2 mL diluted vaccine
• Use a new, sterile needle and syringe
to draw up each new dose.
Information
• If the amount of vaccine remaining
in the vial cannot provide a full dose
of 0.2 mL, discard the vial and any
excess volume.
• Discard any unused vaccine within
12 hours after dilution.
Any unused medicine or waste material should be disposed of in accordance with local
Act
requirements.
1982
7. MEDICINE SCHEDULE
Prescription Medicine.
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 26 of 27
8. SPONSOR
Pfizer New Zealand Limited
P O Box 3998
Auckland, New Zealand
Toll Free Number: 0800 736 363
Released
9. DATE OF FIRST APPROVAL
Date of publication in the New Zealand Gazette of consent to distribute this medicine:
16 December 2021
under
10. DATE OF REVISION OF THE TEXT
27 April 2022
COMIRNATY® is a registered trademark of BioNTech SE. Used under license.
the
Official
Summary of Updates
Section
Update
6.3
Amend shelf life from 9 months to 12 months
6.6
Amend shelf life from 9 months to 12 months
Information
Act 1982
Version: pfdcocii20422
Supersedes: pfdcocii10422
Page 27 of 27
NEW ZEALAND DATA SHEET
1. PRODUCT NAME
COMIRNATY® (orange cap, must dilute), new formulation, 0.1 mg/mL concentrate for
suspension for injection, children 5 to 11 years of age (10 micrograms/0.2 mL dose)
Released
2. QUALITATIVE AND QUANTITATIVE COMPOSITION
This is a multidose vial and
must be diluted before use.
One vial (1.3 mL) contains 10 doses of 0.2 mL after dilution, see Section 4.2 Dose and method
of administration and Section 6.6 Special precautions for disposal and other handling.
under
1 dose (0.2 mL) contains 10 micrograms of tozinameran, a COVID-19 mRNA Vaccine
(embedded in lipid nanoparticles).
Tozinameran is a single-stranded, 5’-capped messenger RNA (mRNA) produced using a cell-
the
free
in vitro transcription from the corresponding DNA templates, encoding the viral spike (S)
protein of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
Official
For the full list of excipients, see Section 6.1 List of excipients.
3. PHARMACEUTICAL FORM
Concentrate for suspension for injection (sterile concentrate).
Information
COMIRNATY is a white to off-white frozen suspension.
4. CLINICAL PARTICULARS
4.1 Therapeutic indications
COMIRNATY (orange cap, must dilute) has provisional consent (see section 5.1) for the
indication below:
Act
Active immunisation to prevent coronavirus disease 2019 (COVID-19) caused by SARS-CoV-
2, in children aged 5 to 11 years.
1982
The use of this vaccine should be in accordance with official recommendations.
4.2 Dose and method of administration
Dose
Children 5 to 11 years of age (i.e. 5 to less than 12 years of age)
COMIRNATY (orange cap, must dilute) is administered intramuscularly as a primary course
of 2 doses (0.2 mL each) at least 21 days apart.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 1 of 32
Booster dose in individuals 5 to 11 years of age
A booster dose of COMIRNATY (orange cap, must dilute) may be administered
intramuscularly at least 6 months after the second dose in individuals 5 to 11 years of age.
Interchangeability
The interchangeability of COMIRNATY with other COVID-19 vaccines to complete the
Released
primary vaccination course has not been established. Individuals who have received 1 dose of
COMIRNATY should receive a second dose of COMIRNATY to complete the primary
vaccination course.
COMIRNATY (orange cap, must dilute) should be used only for children 5 to 11 years of age.
Elderly population
Refer to the Data Sheet for COMIRNATY (grey cap, do not dilute), new formulation, 0.1
under
mg/mL suspension for injection, 12 years of age and older (30 micrograms/dose).
Method of administration
COMIRNATY should be administered intramuscularly, after dilution. The preferred site of
the
administration is the deltoid muscle of the upper arm.
Do not inject COMIRNATY intravascularly, subcutaneously or intradermally.
Official
COMIRNATY should not be mixed in the same syringe with any other vaccines or medicinal
products.
For precautions to be taken before administering COMIRNATY, see Section 4.4 Special
warnings and precautions for use.
Information
COMIRNATY (orange cap, must dilute)
Vials have an orange cap and after
dilution contain ten doses of 0.2 mL of vaccine. In order
to extract ten doses from a single vial, low dead-volume syringes and/or needles should be
used. The low dead-volume syringe and needle combination should have a dead volume of no
more than 35 microlitres. If standard syringes and needles are used, there may not be sufficient
volume to extract a tenth dose from a single vial. Irrespective of the type of syringe and needle:
• Each dose must contain 0.2 mL of vaccine.
Act
• If the amount of vaccine remaining in the vial cannot provide a full dose of 0.2 mL, discard
the vial and any excess volume.
1982
• Do not pool excess vaccine from multiple vials.
For instructions on thawing, handling, dilution and dose preparation of COMIRNATY (orange
cap, must dilute) see Section 6.6 Special precautions for disposal and other handling.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of
excipients.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 2 of 32
4.4 Special warnings and precautions for use
Traceability
In order to improve the traceability of biological medicinal products, the name and the batch
number of the administered product should be clearly recorded.
General recommendations
Released
Hypersensitivity and anaphylaxis
Events of anaphylaxis have been reported. Appropriate medical treatment and supervision
should always be readily available in case of an anaphylactic reaction following the
administration of COMIRNATY.
The individual should be kept under close observation for at least 15 minutes following
vaccination. A second dose of COMIRNATY should not be given to those who have
under
experienced anaphylaxis to the first dose of COMIRNATY.
Myocarditis and pericarditis
Very rare cases of myocarditis and pericarditis have been observed following vaccination with
the
COMIRNATY. These cases have primarily occurred within 14 days following vaccination,
more often after the second vaccination, and more often, but not exclusively in younger men.
There have been reports in females. Based on accumulating data, the reporting rates of
Official
myocarditis and pericarditis after primary series in children ages 5 through <12 years are lower
than in ages 12 through 17 years. Rates of myocarditis and pericarditis in booster doses do not
appear to be higher than after the second dose in the primary series. The cases are generally
mild and individuals tend to recover within a short time following standard treatment and rest.
Cases of myocarditis and pericarditis following vaccination have rarely been associated with
severe outcomes including death.
Information
Healthcare professionals should be alert to the signs and symptoms of myocarditis and
pericarditis, including atypical presentations. Vaccinees should be instructed to seek
immediate medical attention if they develop symptoms indicative of myocarditis or pericarditis
such as (acute and persisting) chest pain, shortness of breath, or palpitations following
vaccination. Non-specific symptoms of myocarditis and pericarditis also include fatigue,
nausea and vomiting, abdominal pain, dizziness or syncope, oedema and cough. Healthcare
professionals should consult guidance and/or specialists to diagnose and treat this condition.
Act
Stress-related responses
Some individuals may have stress-related responses associated with the process of vaccination
1982
itself. Stress-related responses are temporary and resolve on their own. They may include
dizziness, fainting, palpitations, increases in heart rate, alterations in blood pressure, feeling
short of breath, tingling sensations, sweating and/or anxiety. Individuals should be advised to
bring symptoms to the attention of the vaccination provider for evaluation and precautions
should be in place to avoid injury from fainting.
Concurrent illness
Vaccination should be postponed in individuals suffering from acute severe febrile illness or
acute infection. The presence of a minor infection and/or low grade fever should not delay
vaccination.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 3 of 32
Thrombocytopenia and coagulation disorders
As with other intramuscular injections, COMIRNATY should be given with caution in
individuals receiving anticoagulant ther apy or those with thrombocytopenia or any coagulation
disorder (such as haemophilia) bec ause bleeding or bruising may occur following an
intramuscular administration in these individuals.
Immunocompromised individuals
Released
The efficacy, safety and immunogenicity of COMIRNATY has not been assessed in
immunocompromised individuals, including those receiving immunosuppressant therapy. The
efficacy of COMIRNATY may be lower in immunosuppressed individuals.
Duration of protection
The duration of protection afforded by COMIRNATY is unknown as it is still being determined
by ongoing clinical trials.
under
Limitations of vaccine effectiveness
As with any vaccine, vaccination with COMIRNATY may not protect all vaccine recipients.
Individuals may not be fully protected until 7 days after their second dose of COMIRNATY.
the
Use in the elderly
Clinical studies of COMIRNATY include participants 65 years of age and older and their data
Official
contributes to the overall assessment of safety and efficacy. See Section 5.1 Pharmacodynamic
properties, Clinical trials, Efficacy against COVID-19.
Paediatric use
The safety and efficacy of COMIRNATY in children aged less than 5 years of age have not
Information
yet been established.
Effects on laboratory tests
No data available.
4.5 Interactions with other medicines and other forms of interactions
No interaction studies have been performed.
Act
Concomitant administration of COMIRNATY with other vaccines has not been studied.
1982
4.6 Fertility, pregnancy and lactation
Fertility
In a combined fertility and developmental toxicity study, female rats were intramuscularly
administered COMIRNATY prior to mating and during gestation (4 full human doses of
30 micrograms each, spanning between pre-mating day 21 and gestation day 20). SARS-CoV-
2 neutralising antibodies were present in maternal animals from prior to mating to the end of
the study on postnatal day 21 as well as in fetuses and offspring. There were no vaccine related
effects on female fertility and pregnancy rate.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 4 of 32
Pregnancy
There is limited experience with use of COMIRNATY in pregnant women. Animal studies do
not indicate direct or indirect harmful effects with respect to pregnancy, embryo/fetal
development, parturition or post-natal development (see Section 4.6 Fertility, pregnancy and
lactation, Fertility). Administration of COMIRNATY in pregnancy should only be considered
when the potential benefits outweigh any potential risks for the mother and fetus.
Released
Lactation
It is unknown whether tozinameran is excreted in human milk. A combined fertility and
developmental toxicity study in rats did not show harmful effects on offspring development
before weaning (see Section 4.6 Fertility, pregnancy and lactation, Fertility).
4.7 Effects on ability to drive and use machines
under
COMIRNATY has no, or negligible, influence on the ability to drive and use machines.
However, some of the effects mentioned under Section 4.8 Undesirable effects may
temporarily affect the ability to drive or use machines.
the
4.8 Undesirable effects
Summary of safety profile
Official
The safety of COMIRNATY was evaluated in participants 5 years of age and older in 3 clinical
studies that included 24,675 participants (comprised of 22,026 participants 16 years of age and
older, 1,131 adolescents 12 to 15 years of age and 1,518 children 5 to 11 years of age) that have
received at least one dose of COMIRNATY.
Information
Additionally, 306 existing Phase 3 participants at 18 to 55 years of age received a booster dose
of COMIRNATY approximately 6 months after the second dose in the non-placebo-controlled
booster dose portion of Study C4591001. The overall safety profile for the booster dose was
similar to that seen after 2 doses.
In Study C4591031, a placebo-controlled booster study, 5,081 participants 16 years of age and
older were recruited from Study C4591001 to receive a booster dose of COMIRNATY at least
6 months after the second dose. The overall safety profile for the booster dose was similar to
that seen after 2 doses.
Act
In a subset of C4591007 Phase 2/3 participants, 401 participants 5 to 11 years of age received
a booster dose of COMIRNATY at least 5 months after completing the primary series. The
1982
overall safety profile for the booster dose was similar to that seen after the primary series.
Participants 16 years of age and older – after 2 doses
In Study C4591001, a total of 22,026 participants 16 years of age or older received at least 1
dose of COMIRNATY 30 micrograms and a total of 22,021 participants 16 years of age or
older received placebo (including 138 and 145 adolescents 16 and 17 years of age in the
COMIRNATY and placebo groups, respectively). A total of 20,519 participants 16 years of
age or older received 2 doses of COMIRNATY.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 5 of 32
At the time of the analysis of Study C4591001 with a data cut-off of 13 March 2021 for the
placebo-controlled blinded follow-up period up to the participants’ unblinding dates, a total of
25,651 (58.2%) participants (13,031 COMIRNATY and 12,620 placebo) 16 years of age and
older were followed up for ≥4 months after the second dose. This included a total of 15,111
(7,704 COMIRNATY and 7,407 placebo) participants 16 to 55 years of age and a total of
10,540 (5,327 COMIRNATY and 5,213 placebo) participants 56 years and older.
Released
The most frequent adverse reactions in participants 16 years of age and older that received 2
doses were injection site pain (>80%), fatigue (>60%), headache (>50%), myalgia (>40%),
chills (>30%), arthralgia (>20%), pyrexia and injection site swelling (>10%) and were usually
mild or moderate in intensity and resolved within a few days after vaccination. A slightly lower
frequency of reactogenicity events was associated with greater age.
The safety profile in 545 subjects receiving COMIRNATY, that were seropositive for SARS-
CoV-2 at baseline, was similar to that seen in the general population.
under
Study C4591001 also included 200 participants with confirmed stable human
immunodeficiency virus (HIV) infection. The safety profile of the participants receiving
COMIRNATY (n=100) in the individuals with stable HIV infection was similar to that seen in
the general population.
the
Adolescents 12 to 15 years of age – after 2 doses
Official
In an analysis of long term safety follow-up in Study C4591001, 2,260 adolescents
(1,131 COMIRNATY 30 micrograms; 1,129 placebo) were 12 to 15 years of age. Of these,
1,559 adolescents (786 COMIRNATY and 773 placebo) have been followed for ≥ 4 months
after the second dose of COMIRNATY. The safety evaluation in Study C4591001 is ongoing.
The most frequent adverse reactions in adolescents 12 to 15 years of age that received 2 doses
Information
were injection site pain (>90%), fatigue and headache (>70%), myalgia and chills (>40%),
arthralgia and pyrexia (>20%).
Children 5 to 11 years of age – after 2 doses
In an analysis of Study C4591007 Phase 2/3, 2,268 children (1,518 COMIRNATY
10 micrograms; 750 placebo) were 5 to 11 years of age. Of these, 2,158 (95.1%)
(1,444 COMIRNATY 10 micrograms and 714 placebo) children have been followed for at
least 2 months after the second dose. The safety evaluation in Study C4591007 is ongoing.
Act
The most frequent adverse reactions in children 5 to 11 years of age that received 2 doses
included injection site pain (>80%), fatigue (>50%), headache (>30%), injection site redness
and swelling (>20%), myalgia and chills (>10%).
1982
Participants 16 years of age and older – after booster dose
A subset from Study C4591001 Phase 2/3 participants of 306 adults 18 to 55 years of age who
completed the original COMIRNATY 2-dose course, received a booster dose of
COMIRNATY approximately 6 months (range of 4.8 to 8.0 months) after receiving Dose 2.
Of these, 301 participants have been followed for ≥4 months after the booster dose of
COMIRNATY.
The most frequent adverse reactions in participants 18 to 55 years of age were injection site
pain (>80%), fatigue (>60%), headache (>40%), myalgia (>30%), chills and arthralgia (>20%).
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 6 of 32
In Study C4591031, a placebo-controlled booster study, participants 16 years of age and older
recruited from Study C4591001 received a booster dose of COMIRNATY (5,081 participants),
or placebo (5,044 participants) at least 6 months after the second dose of COMIRNATY.
Overall, participants who received a booster dose, had a median follow-up time of 2.8 months
(range 0.3 to 7.5 months) after the booster dose in the blinded placebo-controlled follow-up
period to the cut-off date (8 February 2022). Of these, 1281 participants (895 COMIRNATY
and 386 placebo) have been followed for ≥ 4 months after the booster dose of COMIRNATY.
Released
Children 5 to 11 years of age – after booster dose
In a subset from C4591007, a total of 401 children 5 to 11 years of age received a booster dose
of COMIRNATY 10 mcg at least 5 months (range of 5 to 9 months) after completing the
primary series. The analysis of the C4591007 Phase 2/3 subset is based on data up to the cut-
off date of 22 March 2022 (median follow-up time of 1.3 months).
under
The most frequent adverse reactions in participants 5 to 11 years of age were injection site pain
(>70%), fatigue (>40%), headache (>30%), myalgia, chills, injection site redness, and swelling
(>10%).
Tabulated list of adverse reactions from clinical studies and post-authorisation
the
experience
Adverse reactions observed during clinical studies are listed below according to the following
frequency categories:
Official
Very common (≥ 1/10),
Common (≥ 1/100 to < 1/10),
Uncommon (≥ 1/1,000 to < 1/100),
Information
Rare (≥ 1/10,000 to < 1/1,000),
Very rare (< 1/10,000),
Not known (cannot be estimated from the available data).
Table 1: Adverse reactions from COMIRNATY clinical trials: Individuals 12 years of age
and older
Not known
Rare
Very
Common
Uncommon
(cannot be
System Organ
(≥ 1/10,000 Act
common
(≥ 1/100 to
(≥ 1/1,000 to
estimated from
Class
(≥
to
1/10)
< 1/10)
< 1/100)
the available
< 1/1,000)
data)
1982
Blood and
Lymphadenopathya
lymphatic
system disorders
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 7 of 32
Not known
Rare
Very
Common
Uncommon
(cannot be
System Organ
(≥ 1/10,000
common
(≥ 1/100 to
(≥ 1/1,000 to
estimated from
Class
(≥
to
1/10)
< 1/10)
< 1/100)
the available
< 1/1,000)
data)
Metabolism and
Decreased appetite
nutrition
disorders
Released
Psychiatric
Insomnia
disorders
Nervous system
Headache
Lethargy
Acute
disorders
peripheral
facial
paralysisb
Gastrointestinal
Nausea;
disorders
under
Skin and
Hyperhidrosis;
subcutaneous
Night sweats
tissue disorders
Musculoskeletal
Arthralgia;
and connective
Myalgia the
tissue disorders
General
Injection
Injection
Asthenia; Malaise;
Facial swellingd
disorders and
site pain;
site redness
Official
administration
Fatigue;
site conditions
Chills;
Pyrexiac;
Injection
site
swelling
Information
a A higher frequency of lymphadenopathy (2.8% vs 0.4%) was observed in participants receiving a booster dose
in Study C4591031 compared to participants receiving 2 doses.
b Through the clinical trial safety follow-up period to 14 November 2020, acute peripheral facial paralysis (or
palsy) was reported by four participants in the COMIRNATY group. Onset was Day 37 after Dose 1 (participant
did not receive Dose 2) and Days 3, 9, and 48 after Dose 2. No cases of acute peripheral facial paralysis (or palsy)
were reported in the placebo group.
c A higher frequency of pyrexia was observed after the second dose compared to the first dose. The preferred term
pyrexia is a cluster term covering also body temperature increased..
d Facial swelling in vaccine recipients with a history of injection of dermatological fillers
Act
Table 2.
Adverse Reactions from COMIRNATY clinical trial: Individuals 5 to 11
Years of Age (06 September 2021 Data Cut-off Date)
Rare
1982
Common Uncommon
Not known
Very
≥1/100 to ≥1/1,000 to ≥1/10,000
Very Rare
(cannot be
System Organ
Common
to
<1/10
<1/100
<1/10,000
estimated
Class
≥1/10
<1/1,000
(≥1% to
(≥0.1% to
(<0.01%)
from the
(≥10%)
(≥0.01% to
<10%)
<1%)
available data)
<0.1%)
Blood and
Lymphaden
lymphatic system
opathya
disorders
Immune system
Urticariab,c;
Anaphylaxisb
disorders
Pruritusb,c;
Rashb,c
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 8 of 32
Table 2.
Adverse Reactions from COMIRNATY clinical trial: Individuals 5 to 11
Years of Age (06 September 2021 Data Cut-off Date)
Rare
Common Uncommon
Not known
Very
≥1/100 to ≥1/1,000 to ≥1/10,000
Very Rare
(cannot be
System Organ
Common
to
<1/10
<1/100
<1/10,000
estimated
Class
≥1/10
<1/1,000
(≥1% to
(≥0.1% to
(<0.01%)
from the
(≥10%)
(≥0.01% to
<10%)
<1%)
available data)
Released
<0.1%)
Metabolism and
Decreased
nutrition disorders
appetite
Nervous system
Headache
disorders
Gastrointestinal
Diarrhoeab; Nausea
disorders
Vomitingb
Musculoskeletal
Myalgia
Arthralgia
Pain in
and connective
extremity
under
tissue disorders
(arm)b
General disorders
Injection
Pyrexia
Malaise
and administration site pain;
site conditions
Fatigue;
Chills;
the
Injection
site
swelling;
Official
Injection
site redness
a. A higher frequency of lymphadenopathy was observed in C4591007 (2.5% vs. 0.9%) in
participants receiving a booster dose compared to participants receiving 2 doses.
b. These adverse reactions were identified in the post-authorisation period. The following events
were not reported in participants 5 to 11 Years of Age in Study C4591007 but were reported in
Information
individuals ≥16 years of age in Study C4591001: angioedema, lethargy, asthenia, hyperhidrosis,
and night sweats.
c. The following events are categorised as hypersensitivity reactions: urticaria, pruritus, and rash
Post-marketing experience
Although the events listed in Table 3 were not observed in the clinical trials, they are considered
adverse drug reactions for COMIRNATY as they were reported in the post-marketing
experience. As these reactions were derived from spontaneous reports, the frequencies could
not be determined and are thus considered as not known.
Act
Table 3: Adverse reactions from COMIRNATY post marketing experience
System Organ Class
Adverse Drug Reaction
1982
Immune system disorders
Anaphylaxis
Hypersensitivity reactions (e.g. rash, pruritis, urticaria, angioedema)
Cardiac disorders
Myocarditis
Pericarditis
Nervous system disorders
Dizziness
Gastrointestinal disorders
Diarrhoea
Vomiting
Musculoskeletal and connective
Pain in extremity (arm)a
tissue disorders
General disorders and
Extensive swelling of vaccinated limb
administration site conditions
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 9 of 32
System Organ Class
Adverse Drug Reaction
a A higher frequency of pain in extremity (1.1% vs. 0.8%) was observed in participants receiving a booster dose in
Study C4591031 compared to participants receiving 2 doses.
Reporting suspected adverse effects
Reporting suspected adverse reactions after authorisation of the medicine is important. It allows
continued monitoring of the benefit/risk balance of the medicine. Healthcare professionals are
Released
asked to report any suspected adverse reactions at https://nzphvc.otago.ac.nz/reporting/.
4.9 Overdose
Overdose data is available from 52 study participants included in the clinical trial that due to
an error in dilution received 58 micrograms of COMIRNATY. The COMIRNATY recipients
did not report an increase in reactogenicity or adverse reactions.
under
In the event of overdose, monitoring of vital functions and possible symptomatic treatment is
recommended.
For advice on the management of overdose please contact the National Poisons Centre on 0800
the
POISON (0800 764766).
5. PHARMACOLOGICAL PROPERTIES
Official
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: vaccines, other viral vaccines, ATC code: J07BX03.
Information
Mechanism of action
The nucleoside-modified messenger RNA in COMIRNATY is formulated in lipid
nanoparticles, which enable delivery of the non-replicating RNA into host cells to direct
transient expression of the SARS-CoV-2 spike (S) antigen. The mRNA codes for membrane-
anchored, full-length S with two point mutations within the central helix. Mutation of these
two amino acids to proline locks S in an antigenically preferred prefusion conformation.
COMIRNATY elicits both neutralising antibody and cellular immune responses to the antigen,
which may contribute to protection against COVID-19.
Act
Clinical efficacy and safety
Efficacy
1982
Study C4591001 is a multicentre, multinational, Phase 1/2/3 randomised, placebo-controlled,
observer-blind dose-finding, vaccine candidate selection and efficacy study in participants 12
years of age and older. Randomisation was stratified by age: 12 to 15 years of age, 16 to 55
years of age, or 56 years of age and older, with a minimum of 40% of participants in the ≥56-
year stratum. The study excluded participants who were immunocompromised and those who
had previous clinical or microbiological diagnosis of COVID-19. Participants with pre-existing
stable disease, defined as disease not requiring significant change in therapy or hospitalisation
for worsening disease during the 6 weeks before enrolment, were included as were participants
with known stable infection with HIV, hepatitis C virus (HCV) or hepatitis B virus (HBV).
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 10 of 32
Efficacy in participants 16 years of age and older – after 2 doses
In the Phase 2/3 portion of Study C4591001, based on data accrued through
14 November 2020, approximately 44,000 participants were randomised equally and were to
receive 2 doses of COMIRNATY or placebo. The efficacy analyses included participants that
received their second vaccination within 19 to 42 days after their first vaccination. The majority
(93.1%) of vaccine recipients received the second dose 19 days to 23 days after Dose 1.
Participants are planned to be followed for up to 24 months after Dose 2, for assessments of
Released
safety and efficacy against COVID-19. In the clinical study, participants were required to
observe a minimum interval of 14 days before and after administration of an influenza vaccine
in order to receive either placebo or COMIRNATY. In the clinical study, participants were
required to observe a minimum interval of 60 days before or after receipt of blood/plasma
products or immunoglobulins through to conclusion of the study in order to receive either
placebo or COMIRNATY.
under
The population for the analysis of the primary efficacy endpoint included 36,621 participants
12 years of age and older (18,242 in the COMIRNATY group and 18,379 in the placebo group)
who did not have evidence of prior infection with SARS-CoV-2 through 7 days after the second
dose. In addition, 134 participants were between the ages of 16 to 17 years of age (66 in the
COMIRNATY group and 68 in the placebo group) and 1616 participants 75 years of age and
the
older (804 in the COMIRNATY group and 812 in the placebo group).
At the time of the primary efficacy analysis, participants had been followed for symptomatic
Official
COVID-19 for in total 2,214 person-years for the COMIRNATY group and in total 2,222
person-years for the placebo group.
There were no meaningful clinical differences in overall vaccine efficacy in participants who
were at risk of severe COVID-19 including those with 1 or more comorbidities that increase
the risk of severe COVID-19 (e.g. asthma, body mass index (BMI) ≥30 kg/m2, chronic
Information
pulmonary disease, diabetes mellitus, hypertension).
COMIRNATY efficacy information is presented in Table 4.
Table 4: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2, by age
subgroup – participants without evidence of infection prior to 7 days after Dose 2 –
evaluable efficacy (7 days) population
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
of prior SARS-CoV-2 infection*
Act
COMIRNATY
Placebo
Na = 18,198 Cases
Na = 18,325 Cases
Vaccine efficacy
1982
Subgroup
n1b
n1b
%
Surveillance timec
Surveillance timec
(95% CI)f
(n2d)
(n2d)
All participantse
8
162
95.0
2.214 (17,411)
2.222 (17,511)
(90.0, 97.9)
16 to 64 years
7
143
95.1
1.706 (13,549)
1.710 (13,618)
(89.6, 98.1)
65 years and older
1
19
94.7
0.508 (3848)
0.511 (3880)
(66.7, 99.9)
65 to 74 years
1
14
92.9
0.406 (3074)
0.406 (3095)
(53.1, 99.8)
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 11 of 32
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Na = 18,198 Cases
Na = 18,325 Cases
Vaccine efficacy
Subgroup
n1b
n1b
%
Surveillance timec
Surveillance timec
(95% CI)f
(n2d)
(n2d)
Released
75 years and older
0
5
100.0
0.102 (774)
0.106 (785)
(-13.1, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 [*Case definition: (at least 1 of) fever, new or increased cough,
new or increased shortness of breath, chills, new or increased muscle pain, new loss of taste or smell, sore
throat, diarrhoea or vomiting.]
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the last dose) of
past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not
detected by nucleic acid amplification tests (NAAT) [nasal swab] at Visits 1 and 2), and had negative NAAT
under
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of
the
the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. No confirmed cases were identified in adolescents 12 to 15 years of age.
f. Two-sided confidence interval (CI) for vaccine efficacy (VE) is derived based on the Clopper and Pearson
Official
method adjusted to the surveillance time. CI not adjusted for multiplicity.
In the second primary analysis, efficacy of COMIRNATY in preventing first COVID-19
occurrence from 7 days after Dose 2 compared to placebo was 94.6% (95% credible interval
of 89.9% to 97.3%) in participants 16 years of age and older with or without evidence of prior
infection with SARS-CoV-2.
Information
Additionally, subgroup analyses of the primary efficacy endpoint showed similar efficacy point
estimates across genders, ethnic groups, and participants with medical comorbidities associated
with high risk of severe COVID-19.
Updated efficacy analyses were performed with additional confirmed COVID-19 cases accrued
during blinded placebo-controlled follow-up through 13 March 2021, representing up to
6 months of follow-up after Dose 2 for participants in the efficacy population. Act
The updated vaccine efficacy information is presented in Table 5.
1982
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 12 of 32
Table 5: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2, by age
subgroup – participants without evidence of infection prior to 7 days after Dose 2 –
evaluable efficacy (7 days) population during the placebo-controlled follow-up period
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Na=20,998
Na=21,096
Released
Cases
Cases
n1b
n1b
Surveillance Timec
Surveillance Timec
Vaccine efficacy %
Subgroup
(n2d)
(n2d)
(95% CIe)
All participantsf
77
850
91.3
6.247 (20,712)
6.003 (20,713)
(89.0, 93.2)
16 to 64 years
70
710
90.6
4.859 (15,519)
4.654 (15,515)
(87.9, 92.7)
under
65 years and older
7
124
94.5
1.233 (4192)
1.202 (4226)
(88.3, 97.8)
65 to 74 years
6
98
94.1
0.994 (3350)
0.966 (3379)
(86.6, 97.9)
75 years and older
1
26
96.2
the
0.239 (842)
0.237 (847)
(76.9, 99.9)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
Official
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative
at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
Information
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Two-sided confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method
adjusted to the surveillance time.
f. Included confirmed cases in participants 12 to 15 years of age: 0 in the COMIRNATY group (both without
and with or without evidence of prior SARS-CoV-2 infection); 16 and 18 in the placebo group (without and
with or without evidence of prior SARS-CoV-2 infection, respectively).
Act
Efficacy against severe COVID-19 in participants 12 years of age or older – after 2 doses
As of 13 March 2021, vaccine efficacy against severe COVID-19 is presented only for
participants with or without prior SARS-CoV-2 infection (Table 6) as the COVID-19 case
1982
counts in participants without prior SARS-CoV-2 infection were the same as those in
participants with or without prior SARS-CoV-2 infection in both the COMIRNATY and
placebo groups.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 13 of 32
Table 6. Vaccine Efficacy – First Severe COVID-19 Occurrence in Participants With
or Without* Prior SARS-CoV-2 Infection Based on Food and Drug Administration
(FDA)† Definition After Dose 1 or From 7 Days After Dose 2 in the Placebo-Controlled
Follow-up
COMIRNATY
Placebo
Cases
Cases
Released
n1a
n1a
Vaccine Efficacy %
Surveillance Time (n2b) Surveillance Time (n2b)
(95% CIc)
1
30
96.7
After Dose 1d
8.439e (22,505)
8.288e (22,435)
(80.3, 99.9)
1
21
95.3
7 days after Dose 2f
6.522g (21,649)
6.404g (21,730)
(70.9, 99.9)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at
least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or increased
shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat; diarrhoea;
under
vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative
at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
† Severe illness from COVID-19 as defined by FDA is confirmed COVID-19 and presence of at least 1 of the
the
following:
• Clinical signs at rest indicative of severe systemic illness (respiratory rate ≥30 breaths per minute, heart
rate ≥125 beats per minute, saturation of oxygen ≤93% on room air at sea level, or ratio of arterial
oxygen partial pressure to fractional inspired oxygen <300 mm Hg);
Official
• Respiratory failure [defined as needing high-flow oxygen, noninvasive ventilation, mechanical
ventilation or extracorporeal membrane oxygenation (ECMO)];
• Evidence of shock (systolic blood pressure <90 mm Hg, diastolic blood pressure <60 mm Hg, or
requiring vasopressors);
• Significant acute renal, hepatic, or neurologic dysfunction;
• Admission to an Intensive Care Unit;
Information
• Death.
a. n1 = Number of participants meeting the endpoint definition.
b. n2 = Number of participants at risk for the endpoint.
c. Two-side confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method
adjusted to the surveillance time.
d. Efficacy assessed based on the Dose 1 all available efficacy (modified intention-to-treat) population that
included all randomised participants who received at least 1 dose of study intervention.
e. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from Dose 1 to the end of the surveillance
period.
f. Efficacy assessed based on the evaluable efficacy (7 Days) population that included all eligible randomised
Act
participants who receive all dose(s) of study intervention as randomised within the predefined window, have
no other important protocol deviations as determined by the clinician
g. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
1982
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
Efficacy and immunogenicity in adolescents 12 to 15 years of age – after 2 doses
An analysis of Study C4591001 has been performed in adolescents 12 to 15 years of age up to
a data cutoff date of 13 March 2021.
The vaccine efficacy information in adolescents 12 to 15 years of age is presented in Table 7.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 14 of 32
Table 7: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2 –
participants without evidence of infection and with or without evidence of infection prior
to 7 days after Dose 2 – adolescents 12 to 15 years of age evaluable efficacy (7 days)
population
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
without evidence of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Released
Na = 1005
Na = 978
Cases
Cases
n1b
n1b
Vaccine efficacy
Surveillance timec (n2d)
Surveillance timec (n2d)
% (95% CIe)
Adolescents
0
16
12 to 15 years
0.154 (1001)
0.147 (972)
100.0 (75.3, 100.0)
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
with or without* evidence of prior SARS-CoV-2 infection
under
COMIRNATY
Placebo
Na = 1119
Na = 1110
Cases
Cases
n1b
n1b
Vaccine efficacy
Surveillance timec (n2d)
Surveillance timec (n2d)
% (95% CIe)
the
Adolescents
0
18
12 to 15 years
0.170 (1109)
0.163 (1094)
100.0 (78.1, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at
Official
least 1 symptom consistent with COVID-19 [*Case definition: (at least 1 of) fever, new or increased cough, new
or increased shortness of breath, chills, new or increased muscle pain, new loss of taste or smell, sore throat,
diarrhoea or vomiting).
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the last dose) of
past SARS-CoV-2 infection (i.e, N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not
detected by nucleic acid amplification tests (NAAT) [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
Information
a. N = number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all subjects within each group at
risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of subjects at risk for the endpoint.
e. Confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method adjusted
for surveillance time. CI not adjusted for multiplicity.
In Study C4591001 an analysis of SARS-CoV-2 neutralising titres in a randomly selected
Act
subset of participants was performed to demonstrate non-inferior immune responses (within
1.5-fold) comparing adolescents 12 to 15 years of age to participants 16 to 25 years of age who
had no serological or virological evidence of past SARS-CoV-2 infection. The immune
1982
response to COMIRNATY in adolescents 12 to 15 years of age (n = 190) was non-inferior to
the immune response in participants 16 to 25 years of age (n = 170), based on results for SARS-
CoV-2 neutralising titres at 1 month after Dose 2. The geometric mean titres (GMT) ratio of
the adolescents 12 to 15 years of age group to the participants 16 to 25 years of age group was
1.76, with a 2-sided 95% CI of 1.47 to 2.10, meeting the 1.5-fold non-inferiority criterion (the
lower bound of the 2-sided 95% CI for the geometric mean ratio [GMR] >0.67), which
indicates a statistically greater response in the adolescents 12 to 15 years of age than that of
participants 16 to 25 years of age.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 15 of 32
An updated efficacy analysis of Study C4591001 has been performed in approximately 2,260
adolescents 12 to 15 years of age evaluating confirmed COVID-19 cases accrued up to a data
cut-off date of 2 September 2021, representing up to 6 months of follow-up after Dose 2 for
participants in the efficacy population.
The updated vaccine efficacy information in adolescents 12 to 15 years of age is presented in
Table 8.
Released
Table 8: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Dose 2:
Without Evidence of Infection and With or Without Evidence of Infection
Prior to 7 Days After Dose 2 – Blinded Placebo-Controlled Follow-up Period,
Adolescents 12 To 15 Years of Age Evaluable Efficacy (7 Days) Population
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
without evidence of prior SARS-CoV-2 infection*
under
COMIRNATY
Placebo
Na=1057
Na=1030
Cases
Cases
n1b
n1b
Surveillance Timec
Surveillance Timec
Vaccine Efficacy %
the
(n2d)
(n2d)
(95% CIe)
Adolescents
0
28
100.0
12 to 15 years of age
0.343 (1043)
0.322 (1019)
(86.8, 100.0)
Official
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of
age with or without evidence of prior SARS-CoV-2 infection
COMIRNATY
Placebo
Na=1119
Na=1109
Cases
Cases
n1b
n1b
Information
Surveillance Timec
Surveillance Timec
Vaccine Efficacy %
(n2d)
(n2d)
(95% CIe)
Adolescents
0
30
100.0
12 to 15 years of age
0.362 (1098)
0.345 (1088)
(87.5, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
*
Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum]
negative at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had
negative NAAT (nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the
Act
analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
1982
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each
group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the
end of the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Two-sided confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson
method adjusted for surveillance time.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 16 of 32
Efficacy in children 5 to 11 years of age – after 2 doses
A descriptive efficacy analysis of Study C4591007 has been performed in 1,968 children 5 to
11 years of age without evidence of infection prior to 7 days after Dose 2. This analysis
evaluated confirmed symptomatic COVID-19 cases accrued up to a data cut-off date of 8
October 2021.
The descriptive vaccine efficacy results in children 5 to 11 years of age without evidence of
Released
prior SARS-CoV-2 infection are presented in Table 9. None of the cases accrued met criteria
for severe COVID-19 or multisystem inflammatory syndrome in children (MIS-C). No cases
of COVID-19 were observed in either the vaccine group or the placebo group in participants
with evidence of prior SARS-CoV-2 infection.
Table 9: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Dose 2:
Without Evidence of Infection Prior to 7 Days After Dose 2 – Phase 2/3 – Children 5 To
under
11 Years of Age Evaluable Efficacy Population
First COVID-19 occurrence from 7 days after Dose 2 in children 5 to 11 years of age
without evidence of prior SARS-CoV-2 infection*
COMIRNATY±
the
10 micrograms/dose
Placebo
Na=1305
Na=663
Cases
Cases
n1b
n1b
Official
Surveillance Timec
Surveillance Timec
Vaccine Efficacy %
(n2d)
(n2d)
(95% CI)
Children 5 to
3
16
90.7
11 years of age
0.322 (1273)
0.159 (637)
(67.7, 98.3)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
Information
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
*
Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum]
negative at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had
negative NAAT (nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the
analysis.
± Pfizer-BioNTech COVID-19 Vaccine (10 micrograms modRNA).
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each
Act
group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the
end of the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
1982
Immunogenicity in children 5 to 11 years of age – after 2 doses
Study C4591007 is a Phase 1/2/3 study comprised of an open-label vaccine dose-finding
portion (Phase 1) and a multicentre, multinational, randomised, saline placebo-controlled,
observer-blind efficacy portion (Phase 2/3) that has enrolled participants 5 to 11 years of age.
In C4591007, an analysis of SARS-CoV-2 50% neutralising titres (NT50) 1 month after Dose
2 in a randomly selected subset of participants demonstrated effectiveness by immunobridging
of immune responses comparing children 5 to 11 years of age in the Phase 2/3 part of Study
C4591007 to participants 16 to 25 years of age in the Phase 2/3 part of Study C4591001 who
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 17 of 32
had no serological or virological evidence of past SARS-CoV-2 infection up to 1 month after
Dose 2, meeting the prespecified immunobridging criteria for both the geometric mean ratio
(GMR) and the seroresponse difference with seroresponse defined as achieving at least 4-fold
rise in SARS-CoV-2 NT50 from baseline (before Dose 1).
The ratio of the SARS-CoV-2 NT50 in children 5 to 11 years of age to that of young adults 16
to 25 years of age was 1.04 (2-sided 95% CI: 0.93, 1.18), as presented in Table 10.
Released
Table 10:
Summary of geometric mean ratio for 50% neutralising titre – Comparison
of children 5 to 11 years of age (Study C4591007) to participants 16 to 25 years of age
(Study C4591001) – participants without* evidence of infection up to 1 month after Dose
2 – evaluable immunogenicity population
COMIRNATY
5 to 11 years/
10 microgram/dose 30 microgram/dose
16 to 25 years
5 to 11 years
16 to 25 years
under
na=264
na=253
Met
Time
GMTc
GMTc
GMRd
immunobridging
Assay
pointb
(95% CIc)
(95% CIc)
(95% CId)
objectivee
(Y/N)
the
SARS-CoV-2
neutralisation 1 month
1197.6
1146.5
1.04
Y
assay - NT50 after
(1106.1, 1296.6)
(1045.5, 1257.2)
(0.93, 1.18)
Official
(titre)f
Dose 2
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre;
LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test; NT50 = 50% neutralising titre;
SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
*Participants who had no serological or virological evidence (up to 1 month post-Dose 2 blood sample collection)
of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and 1 month after Dose
Information
2, SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2, and negative NAAT (nasal swab) at
any unscheduled visit up to 1 month after Dose 2 blood collection) and had no medical history of COVID-
19 were included in the analysis.
a. n = Number of participants with valid and determinate assay results for the specified assay at the given
dose/sampling time point.
b. Protocol-specified timing for blood sample collection.
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
d. GMRs and 2-sided 95% CIs were calculated by exponentiating the mean difference of the logarithms of the
titres (Group 1[5 to 11 years of age] - Group 2 [16 to 25 years of age]) and the corresponding CI (based on
Act
the Student t distribution).
e. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67 and
the point estimate of the GMR is ≥0.8.
f. SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation
1982
Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus
neutralisation is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution
at which 50% of the virus is neutralised.
Among participants without prior evidence of SARS-CoV-2 infection up to 1 month after
Dose 2, 99.2% of children 5 to 11 years of age and 99.2% of participants 16 to 25 years of age
had a seroresponse from before vaccination to 1 month after Dose 2. The difference in
proportions of participants who had seroresponse between the 2 age groups (children – young
adult) was 0.0% (2-sided 95% CI: -2.0%, 2.2%) as presented in Table 11.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 18 of 32
Table 11: Difference in percentages of participants with seroresponse – participants
without evidence of infection up to 1 month after Dose 2 – immunobridging subset –
Phase 2/3 – comparison of 5 to 11 years of age to Study C4591001 Phase 2/3 16 to 25 years
of age – evaluable immunogenicity population
COMIRNATY
10
30
5 to 11 years/
microgram/dose microgram/dose
Released
16 to 25 years
5 to 11 years
16 to 25 years
Na=264
Na=253
Met
Time
nc (%)
nc (%)
Difference %e immunobridging
Assay
pointb
(95% CId)
(95% CId)
(95% CIf)
objectiveg
(Y/N)
SARS-CoV-2
1 month
neutralisation
262 (99.2)
251 (99.2)
0.0
under
after
Y
assay – NT50
(97.3, 99.9)
(97.2, 99.9)
(-2.0, 2.2)
Dose 2
(titre)h
Abbreviations: LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test;
N-binding = SARS-CoV-2 nucleoprotein–binding; NT50 = 50% neutralising titre 50; SARS-CoV-2 = severe acute
respiratory syndrome coronavirus 2.
Note: Seroresponse is defined as achieving a ≥4
the -fold rise from baseline (before Dose 1). If the baseline measurement
is below the LLOQ, a postvaccination assay result ≥4 × LLOQ is considered a seroresponse.
Note: Participants who had no serological or virological evidence (up to 1 month post-Dose 2 blood sample
collection) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and 1 month after
Official
Dose 2, SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2, and negative NAAT (nasal swab) at
any unscheduled visit up to 1 month after Dose 2 blood collection) and had no medical history of COVID-19 were
included in the analysis.
a. N = number of participants with valid and determinate assay results both before vaccination and at 1 month after
Dose 2. These values are the denominators for the percentage calculations.
b. Protocol-specified timing for blood sample collection.
c. n = Number of participants with seroresponse for the given assay at the given dose/sampling time point.
Information
d. Exact 2-sided CI based on the Clopper and Pearson method.
e. Difference in proportions, expressed as a percentage (Group 1 [5 to 11 years of age] – Group 2 [16 to 25 years
of age]).
f. 2-Sided CI, based on the Miettinen and Nurminen method for the difference in proportions, expressed as a
percentage.
g. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the difference in proportions is greater
than -10.0%.
h. SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation Assay.
The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus neutralisation is
read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution at which 50% of
Act
the virus is neutralised.
Immunogenicity in participants 18 years of age and older – after booster dose
1982
Effectiveness of a booster dose of COMIRNATY was based on an assessment of 50%
neutralising titres (NT50) against SARS-CoV-2 (USA_WA1/2020). In Study C4591001,
analyses of NT50 1 month after the booster dose compared to 1 month after the primary series
in individuals 18 to 55 years of age who had no serological or virological evidence of past
SARS-CoV-2 infection up to 1 month after the booster vaccination demonstrated
noninferiority for both GMR and difference in seroresponse rates. Seroresponse for a
participant was defined as achieving a ≥4-fold rise in NT50 from baseline (before Dose 1),
These analyses are summarised in Table 12.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 19 of 32
Table 12. SARS-CoV-2 neutralisation assay - NT50 (titre)† (SARS-CoV-2
USA_WA1/2020) – GMT and seroresponse rate comparison of 1 month after booster dose
to 1 month after primary series – participants 18 to 55 years of age without evidence of
infection up to 1 month after booster dose* – booster dose evaluable immunogenicity
population±
1 month after
booster dose/-
Released
1 month after
Met
1 month after
1 month after
primary
noninferiority
booster dose
primary series
series
objective
n
(95% CI)
(95% CI)
(97.5% CI)
(Y/N)
Geometric mean
50% neutralising
2466.0b
755.7
b
3.26c
titre (GMTb)
212a
(2202.6, 2760.8)
(663.1, 861.2)
(2.76, 3.86)
Yd
Seroresponse rate
199f
190f
under
(%) for 50%
99.5%
95.0%
4.5%g
neutralising titre†
200e
(97.2%, 100.0%) (91.0%, 97.6%)
(1.0%, 7.9%
h)
Yi
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre;
LLOQ = lower limit of quantitation; N-binding = SARS-CoV-2 nucleoprotein-binding; NAAT = nucleic acid
amplification test; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute respiratory syndrome
coronavirus 2; Y/N = yes/no.
the
†
SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation
Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus
neutralisation is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum
Official
dilution at which 50% of the virus is neutralised.
*
Participants who had no serological or virological evidence (up to 1 month after receipt of a booster dose
of Comirnaty) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative and
SARS-CoV-2 not detected by NAAT [nasal swab]) and had a negative NAAT (nasal swab) at any
unscheduled visit up to 1 month after the booster dose were included in the analysis.
± All eligible participants who had received 2 doses of Comirnaty as initially randomised, with Dose 2
received within the predefined window (within 19 to 42 days after Dose 1), received a booster dose of
Information
Comirnaty, had at least 1 valid and determinate immunogenicity result after booster dose from a blood
collection within an appropriate window (within 28 to 42 days after the booster dose), and had no other
important protocol deviations as determined by the clinician.
a. n = Number of participants with valid and determinate assay results at both sampling time points within
specified window.
b. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
c. GMRs and 2-sided 97.5% CIs were calculated by exponentiating the mean differences in the logarithms of
the assay and the corresponding CIs (based on the Student t distribution).
d. Noninferiority is declared if the lower bound of the 2-sided 97.5% CI for the GMR is > 0.67 and the point
Act
estimate of the GMR is ≥ 0.80.
e. n = Number of participants with valid and determinate assay results for the specified assay at baseline,
1 month after Dose 2 and 1 month after the booster dose within specified window. These values are the
1982
denominators for the percentage calculations.
f. Number of participants with seroresponse for the given assay at the given dose/sampling time point. Exact
2-sided CI based on the Clopper and Pearson method.
g. Difference in proportions, expressed as a percentage (1 month after booster dose – 1 month after Dose 2).
h. Adjusted Wald 2-sided CI for the difference in proportions, expressed as a percentage.
i. Noninferiority is declared if the lower bound of the 2-sided 97.5% CI for the percentage difference is
> -10%.
Relative vaccine efficacy in participants 16 years of age and older – after booster dose
An interim efficacy analysis of Study C4591031, a placebo-controlled booster study, was
performed in approximately 10,000 participants 16 years of age and older who were recruited
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 20 of 32
from Study C4591001, evaluated confirmed COVID-19 cases accrued from at least 7 days after
booster vaccination up to a data cut-off date of 8 February 2022 (a period when Delta and then
Omicron was the predominant variant), which represents a median of 2.8 months (range 0.3 to
7.5 months) post-booster follow-up. Vaccine efficacy of the COMIRNATY booster dose after
the primary series relative to the placebo booster group who only received the primary series
dose was assessed. The relative vaccine efficacy information for participants 16 years of age
and older is presented in Table 13.
Released
Table 13: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Booster
Vaccination – Participants 16 Years of Age and Older Without Evidence of
Infection and Participants With or Without Evidence of Infection Prior to 7
Days After Booster Vaccination – Evaluable Efficacy Population
First COVID-19 occurrence from 7 days after booster dose in participants without evidence
of prior SARS-CoV-2 infection*
under
COMIRNATY
Placebo
Na=4689
Na=4664
Cases
Cases
n1b
n1b
Relative Vaccine
Surveillance Timec
Surveillance Timec
Efficacye %
the
(n2d)
(n2d)
(95% CIf)
First COVID-19
occurrence from
Official
7 days after booster
63
148
63.9
vaccination
1.098 (4639)
0.932 (4601)
(51.1, 73.5)
First COVID-19 occurrence from 7 days after booster dose in participants with or without
evidence of prior SARS-CoV-2 infection
COMIRNATY
Placebo
Na=4997
Na=4942
Information
Cases
Cases
n1b
n1b
Relative Vaccine
Surveillance Timec
Surveillance Timec
Efficacye %
(n2d)
(n2d)
(95% CIf)
First COVID-19
occurrence from
7 days after booster
67
150
62.4
vaccination
1.179 (4903)
0.989 (4846)
(49.5, 72.2)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
Act
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhea; vomiting).
*
Participants who had no serological or virological evidence (prior to 7 days after receipt of the booster
1982
vaccination) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and
SARS-CoV-2 not detected by NAAT [nasal swab] at Visit 1, and had a negative NAAT [nasal swab] at
any unscheduled visit prior to 7 days after booster vaccination) were included in the analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each
group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after the booster
vaccination to the end of the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Relative vaccine efficacy of the COMIRNATY booster group relative to the placebo group (non-booster).
f. Two-sided confidence interval (CI) for relative vaccine efficacy is derived based on the Clopper and
Pearson method adjusted for surveillance time.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 21 of 32
Immunogenicity in children 5 to 11 years of age – after booster dose
Effectiveness of a booster dose of COMIRNATY was based on an assessment of NT50 against
the reference strain of SARS-CoV-2 (USA_WA1/2020). Analyses of NT50 1 month after the
booster dose compared to before the booster dose demonstrated a substantial increase in GMTs
in individuals 5 to 11 years of age who had no serological or virological evidence of past
SARS-CoV-2 infection up to 1 month after the booster dose. This analysis is summarised in
Table 14.
Released
Table 14: Summary of Geometric Mean Titres – NT50 – Participants Without Evidence
of Infection – Phase 2/3 – Immunogenicity Set – 5 to 11 Years of Age –
Evaluable Immunogenicity Population
COMIRNATY 10 mcg/Dose
3-Dose Set
2-Dose Set
Total
under
Dose/
Sampling
GMTc
GMTc
GMTc
Assay
Time Pointa
nb
(95% CIc)
nb
(95% CIc)
nb
(95% CIc)
20.5
20.5
20.5
1 month Prevax
79
(20.5, 20.5)
67
(20.5, 20.5) 146
(20.5, 20.5)
the
SARS-CoV-2 1 month after Dose
1659.4
1110.7
1253.9
neutralisation
2
29 (1385.1, 1988.0) 67 (965.3, 1278.1) 96 (1116.0, 1408.9)
Official
assay - NT50
271.0
271.0
(titre)
3 months Prevax
67
(229.1, 320.6)
-
-
67 (229.1, 320.6)
1 month after Dose
2720.9
2720.9
3
67 (2280.1, 3247.0) -
-
67 (2280.1, 3247.0)
Abbreviations: CI = confidence interval; GMT = geometric mean titre; LLOQ = lower limit of quantitation;
NAAT = nucleic acid amplification test; N-binding = SARS-CoV-2 nucleoprotein–binding; NT50 = 50%
Information
neutralising titre; Prevax = before vaccination; SARS-CoV-2 = severe acute respiratory syndrome coronavirus
2.
Note: Three-dose immunogenicity set included the first 130 participants who received Dose 3 and completed
1-month post–Dose 3 visit prior to March 15, 2022. Among those, 30 had blood sample collection at 1-month
post-Dose 2. Two-dose immunogenicity set included an extra 67 participants randomly selected from previous
Dose-2 evaluable immunogenicity population and without evidence of infection up to 1-month post–Dose 2
subset used for 2-dose immunobridging analysis.
Note: Participants included in this analysis had no serological or virological evidence of past SARS-CoV-2
infection up to the 1-month post–Dose 2 (for 1-month post–Dose 2 time point) or 1-month post–Dose 3 (for pre–
Dose 3 and 1-month post–Dose 3 time point) study blood sample collection. Having no evidence of past SARS-
CoV-2 infection up to 1-month post–Dose 2 was defined as having a negative N-binding antibody (serum) result
Act
at the Dose 1 and 1-month post–Dose 2 study visits; a negative NAAT (nasal swab) result at the Dose 1 and
Dose 2 study visits and any unscheduled visit prior to the 1-month post–Dose 2 blood sample collection; and no
medical history of COVID-19. Having no evidence of past SARS-CoV-2 infection up to 1-month post-Dose 3
1982
was defined as having a negative N-binding antibody (serum) result at the Dose 1, 1-month post–Dose 2 (if
available), Dose 3, and 1-month post–Dose 3 study visits; a negative NAAT (nasal swab) result at the Dose 1,
Dose 2, and Dose 3 study visits and any unscheduled visit prior to the 1-month post–Dose 3 blood sample
collection; and no medical history of COVID-19.
a. Protocol-specified timing for blood sample collection.
b. n = Number of participants with valid and determinate assay results for the specified assay at the given
dose/sampling time point.
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 ×
LLOQ.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 22 of 32
Immunogenicity in children 5 to 11 years of age on the Omicron variant (B1.1.529) –
after booster dose
The neutralising GMTs against both the Omicron variant (B1.1.529) and reference strain were
substantially increased after booster vaccination compared with after the 2-dose primary series.
At 1-month post-Dose 2, the observed neutralising GMTs for the Omicron variant (B1.1.529)
and reference strain were 27.6 and 323.8, respectively. At 1-month post-Dose 3, the observed
neutralising GMTs for the Omicron variant (B1.1.529) and reference strain were 614.4 and
Released
1702.8, respectively (see Table 15).
For the Omicron variant (B1.1.529), neutralising titres after booster vaccination (1-month post-
Dose 3) increased 22-fold over those after the 2-dose primary series (1-month post-Dose 2).
For the reference strain, the increase after the booster relative to the primary series was 5.3-
fold.
under
Table 15: Summary of Geometric Mean Titres – Omicron-Neutralisation Subset –
Participants Without Evidence of Infection – Phase 2/3 – Immunogenicity Set
– 5 to 11 Years of Age – Evaluable Immunogenicity Population
COMIRNATY
the
10 mcg/Dose
Vaccine Group (as Randomised)
GMTc
Official
Assay
Time Pointb
nb
(95% CIc)
SARS-COV-2 FFRNT-
27.6
B.1.1.529 strain
1 month after Dose 2
29
(22.1, 34.5)
(Omicron) - NT50
614.4
(titre)
1 month after Dose 3
17
(410.7, 919.2)
323.8
SARS-CoV-2 FFRNT-
Information
1 month after Dose 2
29
(267.5, 392.1)
reference strain - NT50
1702.8
(titre)
1 month after Dose 3
17
(1282.6, 2260.7)
Abbreviations: CI = confidence interval; FFRNT = fluorescence focus reduction neutralisation test;
GMT = geometric mean titre; LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test;
N-binding = SARS-CoV-2 nucleoprotein–binding; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute
respiratory syndrome coronavirus 2.
Note: Participants included in this analysis had no serological or virological evidence of past SARS-CoV-2
infection up to the 1-month post–Dose 2 (for 1-month post–Dose 2 time point) or 1-month post–Dose 3 (for
1-month post–Dose 3 time point) study blood sample collection. Having no evidence of past SARS-CoV-2
Act
infection up to 1-month post–Dose 2 was defined as having a negative N-binding antibody (serum) result at
the Dose 1 and 1-month post–Dose 2 study visits; a negative NAAT (nasal swab) result at the Dose 1 and Dose
2 study visits and any unscheduled visit prior to the 1-month post–Dose 2 blood sample collection; and no
medical history of COVID-19. Having no evidence of past SARS-CoV-2 infection up to 1-month post–Dose 3
1982
was defined as having a negative N-binding antibody (serum) result at the Dose 1, 1-month post–Dose 2 (if
available), Dose 3, and 1-month post–Dose 3 study visits; a negative NAAT (nasal swab) result at the Dose 1,
Dose 2, and Dose 3 study visits and any unscheduled visit prior to the 1-month post–Dose 3 blood sample
collection; and no medical history of COVID-19.
a. Protocol-specified timing for blood sample collection.
b. n = Number of participants with valid and determinate assay results for the specified assays at the given
dose/sampling time point.
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 23 of 32
This medicine has been given a provisional consent under Section 23 of the Act. This means
that further evidence on this medicine is awaited or that there are specific conditions of use.
Refer to the consent notice published in the New Zealand Gazette for the specific conditions.
5.2 Pharmacokinetic properties
Not applicable.
Released
5.3 Preclinical safety data
Genotoxicity/Carcinogenicity
Neither genotoxicity nor carcinogenicity studies were performed. The components of
COMIRNATY (lipids and mRNA) are not expected to have genotoxic potential.
under
6. PHARMACEUTICAL PARTICULARS
6.1 List of excipients the
((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
Official
Distearoylphosphatidylcholine (DSPC)
Cholesterol
Trometamol
Trometamol hydrochloride
Information
Sucrose
Water for injections
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those
mentioned in Section 6.6 Special precautions for disposal and other handling. Act
6.3 Shelf life
1982
COMIRNATY (orange cap, must dilute)
Unopened vial
Frozen vial
18 months when stored at -90°C to -60°C.
The vaccine will be received frozen at -90°C to -60°C. Frozen vaccine can be stored either at
-90°C to -60°C or 2°C to 8°C upon receipt.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 24 of 32
When stored frozen at -90°C to -60°C, 10-vial packs of the vaccine can be thawed at 2°C to 8°C
for 4 hours or individual vials can be thawed at room temperature (up to 30°C) for 30 minutes.
Thawed vial
If the vaccine is received at 2°C to 8°C it should be stored at 2°C to 8°C. Once removed from
frozen storage, the unopened vial may be stored refrigerated at 2°C to 8°C for a single period
of up to 10 weeks within the 18-month shelf life.
Released
Upon moving the product to 2°C to 8°C storage, the updated expiry date must be written on
the outer carton and the vaccine should be used or discarded by the updated expiry date. The
original expiry date should be crossed out.
Check that the expiry date on the outer carton has been updated to reflect the refrigerated expiry
date and that the original expiry date has been crossed out.
under
When stored frozen at -90°C to -60°C, the vaccine can be thawed at either 2°C to 8°C or at
temperatures up to 30°C.
Prior to use, the unopened vials can be stored for up to 12 hours at temperatures between
8ºC to 30ºC.
the
Thawed vials can be handled in room light conditions.
Official
Once thawed COMIRNATY (orange cap, must dilute) should not be re-frozen.
Diluted medicinal product
Chemical and physical in-use stability has been demonstrated for 12 hours at 2ºC to 30°C, after
dilution with sodium chloride 9 mg/mL (0.9%) solution for injection. From a microbiological
Information
point of view, unless the method of dilution precludes the risk of microbial contamination, the
product should be used immediately. If not used immediately, in-use storage times and
conditions are the responsibility of the user.
6.4 Special precautions for storage
COMIRNATY (orange cap, must dilute) can be stored in a refrigerator at 2°C to 8°C for a
single period of up to 10 weeks, not exceeding the original expiry date (EXP). The expiry date
for storage at -90°C to -60°C is printed on the vial and outer carton after “EXP”.
Act
Check that the expiry date has been updated to reflect the refrigerated EXP date and that the
original expiry date has been crossed out.
1982
Store in the original package to protect from light. During storage, minimise exposure to room
light, and avoid exposure to direct sunlight and ultraviolet light.
For detailed instructions see Section 6.6 Special precautions for disposal and other handling.
Once thawed, the vaccine cannot be re-frozen.
Thawed vials can be handled in room light conditions.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 25 of 32
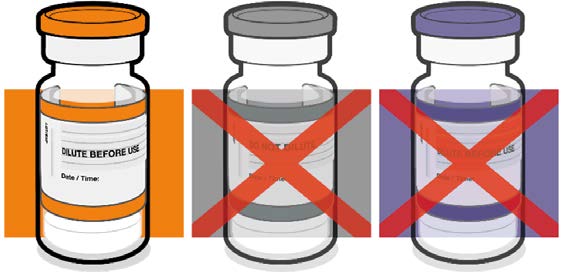


For storage conditions after thawing and dilution of the medicinal product, see Section 6.3
Shelf life.
For additional advice on storing COMIRNATY, contact Pfizer New Zealand on 0800 736 363.
6.5 Nature and contents of container
Released
COMIRNATY (orange cap, must dilute) 1.3 mL fill volume in 2 mL clear multidose vial
(Type I glass) with a stopper (synthetic bromobutyl rubber) and an orange flip-off plastic cap
with aluminium seal. Each vial contains 10 doses, see Section 6.6 Special precautions for
disposal and other handling.
Pack size: 10 vials, 195 vials
Not all pack sizes may be marketed.
under
6.6 Special precautions for disposal and other handling
COMIRNATY (orange cap, must dilute)
the
The vaccine should be prepared by a healthcare professional using aseptic technique to ensure
the sterility of the prepared diluted suspension.
Official
COMIRNATY (orange cap, must dilute)
Dose Verification
• Verify that the vial has an orange
Information
plastic cap.
Orange cap
• Only the orange cap vial can be used
for children age 5 to 11 years.
10 micrograms
Act 1982
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 26 of 32
 COMIRNATY (orange cap, must dilute)
Handling Prior To Use
COMIRNATY (orange cap, must dilute)
Handling Prior To Use
• If the multidose vial is stored frozen
it must be thawed prior to use.
Frozen vials should be transferred to
Released
an environment of 2°C to 8°C to
thaw; a 10 vial pack may take
4 hours to thaw. Ensure vials are
Store for up to
completely thawed prior to use.
10 weeks at
• Unopened vials can be stored for up
2 °C to 8 °C
to 10 weeks at 2°C to 8°C within the
18 month shelf life.
•
under
Alternatively, individual frozen vials
may be thawed for 30 minutes at
temperatures up to 30°C for
immediate use.
the Official
Information
Act 1982
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 27 of 32
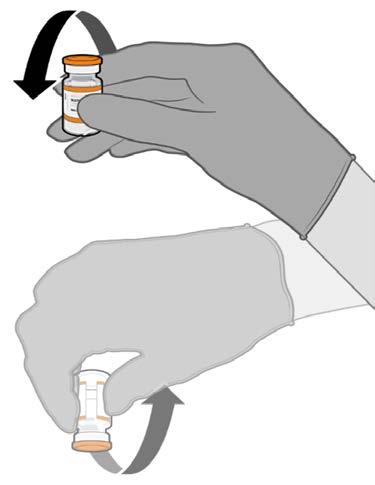
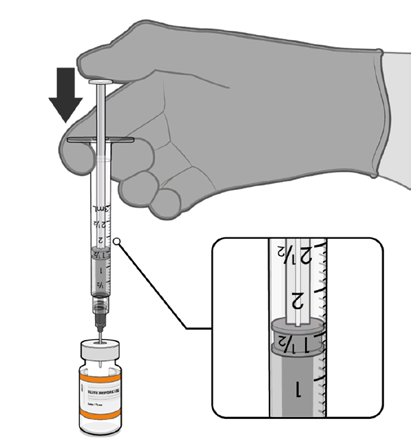 COMIRNATY (orange cap, must dilute)
Mixing Prior To Dilution
COMIRNATY (orange cap, must dilute)
Mixing Prior To Dilution
• Allow the thawed vial to come to
room temperature and gently invert it
10 times prior to dilution. Do not
Released
shake.
• Prior to dilution, the thawed
suspension may contain white to off-
white opaque amorphous particles.
under
the
Official
Dilution
• The thawed vaccine must be diluted
in its original vial with 1.3 mL
sodium chloride 9 mg/mL (0.9%)
solution for injection, using a
Information
21 gauge or narrower needle and
aseptic techniques.
Act 1982
1.3 mL of 0.9% sodium chloride
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 28 of 32
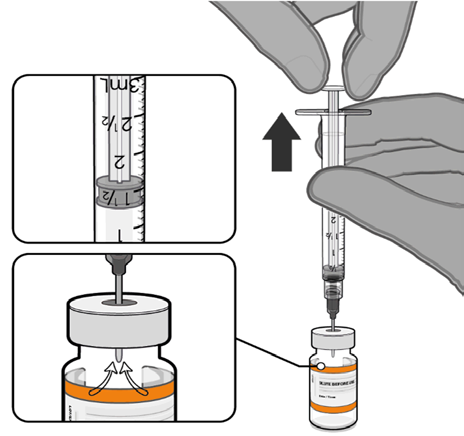
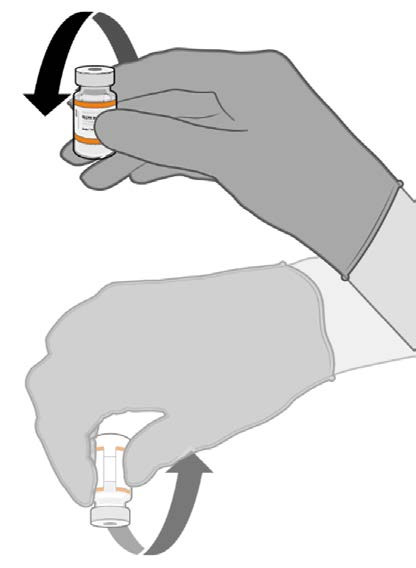 COMIRNATY (orange cap, must dilute)
Dilution (continued)
COMIRNATY (orange cap, must dilute)
Dilution (continued)
• Equalise vial pressure before
removing the needle from the vial
stopper by withdrawing 1.3 mL air
Released
into the empty diluent syringe.
under
the
Pull back plunger to 1.3 mL to
remove air from vial.
Official
• Gently invert the diluted suspension
10 times. Do not shake.
• The diluted vaccine should present as
a white to off-white suspension with
no particulates visible. Do not use
Information
the diluted vaccine if particulates or
discoloration are present.
Act 1982
Gently × 10
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 29 of 32
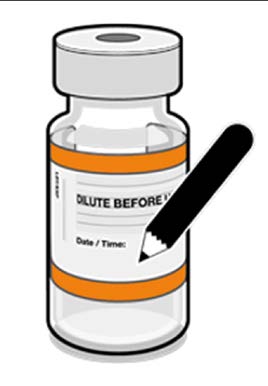 COMIRNATY (orange cap, must dilute)
Dilution (continued)
COMIRNATY (orange cap, must dilute)
Dilution (continued)
• The diluted vials should be marked
with the appropriate date and time.
• After dilution, store at 2°C to 30°C
Released
and use within 12 hours.
• Do not freeze or shake the diluted
dispersion. If refrigerated, allow the
diluted suspension to come to room
temperature prior to use.
under
Record appropriate date and time.
Use within 12 hours after dilution.
the
Official
Information
Act 1982
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 30 of 32
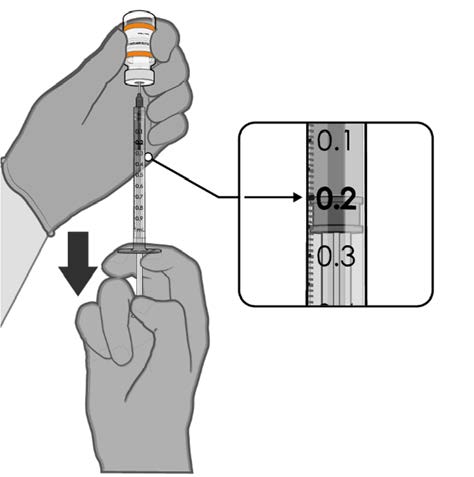 COMIRNATY (orange cap, must dilute)
Preparation of Individual 0.2 mL Doses of COMIRNATY (orange cap, must dilute)
COMIRNATY (orange cap, must dilute)
Preparation of Individual 0.2 mL Doses of COMIRNATY (orange cap, must dilute)
• Using aseptic technique, cleanse the
vial stopper with a single-use
antiseptic swab.
Released
• Withdraw 0.2 mL of COMIRNATY
(orange cap, must dilute).
Low dead-volume syringes and/or
needles should be used in order to
extract 10 doses from a single vial.
The low dead-volume syringe and
needle combination should have a
under
dead volume of no more than
35 microlitres.
If standard syringes and needles are
the
used, there may not be sufficient
volume to extract ten doses from a
single vial.
•
Official Each dose must contain 0.2 mL of
vaccine.
• Discard syringe and needle after
administration to a single patient.
0.2 mL diluted vaccine
• Use a new, sterile needle and syringe
to draw up each new dose.
Information
• If the amount of vaccine remaining
in the vial cannot provide a full dose
of 0.2 mL, discard the vial and any
excess volume.
• Discard any unused vaccine within
12 hours after dilution.
Any unused medicine or waste material should be disposed of in accordance with local
Act
requirements.
1982
7. MEDICINE SCHEDULE
Prescription Medicine.
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 31 of 32
8. SPONSOR
Pfizer New Zealand Limited
P O Box 3998
Auckland, New Zealand
Toll Free Number: 0800 736 363
Released
9. DATE OF FIRST APPROVAL
Date of publication in the New Zealand Gazette of consent to distribute this medicine:
16 December 2021
under
10. DATE OF REVISION OF THE TEXT
07 December 2022
COMIRNATY® is a registered trademark of BioNTech SE. Used under license.
the
Official
Summary of Updates
Section
Update
6.3
Update shelf life from 12 months to 18 months.
Information
6.6
Update shelf life from 12 months to 18 months.
Act 1982
Version: pfdcocii21222
Supersedes: pfdcocii11222
Page 32 of 32
NEW ZEALAND DATA SHEET
1. PRODUCT NAME
COMIRNATY® (orange cap, must dilute), new formulation, 0.1 mg/mL concentrate for
suspension for injection, children 5 to 11 years of age (10 micrograms/0.2 mL dose)
Released
2. QUALITATIVE AND QUANTITATIVE COMPOSITION
This is a multidose vial and
must be diluted before use.
One vial (1.3 mL) contains 10 doses of 0.2 mL after dilution, see Section 4.2 Dose and method
of administration and Section 6.6 Special precautions for disposal and other handling.
under
1 dose (0.2 mL) contains 10 micrograms of tozinameran, a COVID-19 mRNA Vaccine
(embedded in lipid nanoparticles).
Tozinameran is a single-stranded, 5’-capped messenger RNA (mRNA) produced using a cell-
free
in vitro transcription from the corresponding DNA templates, encoding the viral spike (S)
the
protein of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
For the full list of excipients, see Section 6.1 List of excipients.
Official
3. PHARMACEUTICAL FORM
Concentrate for suspension for injection (sterile concentrate).
Information
COMIRNATY is a white to off-white frozen suspension.
4. CLINICAL PARTICULARS
4.1 Therapeutic indications
COMIRNATY (orange cap, must dilute) has provisional consent (see section 5.1) for the
indication below:
Act
Active immunisation to prevent coronavirus disease 2019 (COVID-19) caused by SARS-CoV-
2, in children aged 5 to 11 years.
1982
The use of this vaccine should be in accordance with official recommendations.
4.2 Dose and method of administration
Dose
Children 5 to 11 years of age (i.e. 5 to less than 12 years of age)
COMIRNATY (orange cap, must dilute) is administered intramuscularly as a primary course
of 2 doses (0.2 mL each) at least 21 days apart.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 1 of 32
Booster dose in individuals 5 to 11 years of age
A booster dose of COMIRNATY (orange cap, must dilute) may be administered
intramuscularly at least 6 months after the second dose in individuals 5 to 11 years of age.
Interchangeability
The interchangeability of COMIRNATY with other COVID-19 vaccines to complete the
primary vaccina
Released tion course has not been established. Individuals who have received 1 dose of
COMIRNATY should receive a second dose of COMIRNATY to complete the primary
vaccination course.
COMIRNATY (orange cap, must dilute) should be used only for children 5 to 11 years of age.
Elderly population
Refer to the Data Sheet for COMIRNATY (grey cap, do not dilute), new formulation, 0.1
under
mg/mL suspension for injection, 12 years of age and older (30 micrograms/dose).
Method of administration
COMIRNATY should be administered intramuscularly, after dilution. The preferred site of
administration is the deltoid muscle of
the the upper arm.
Do not inject COMIRNATY intravascularly, subcutaneously or intradermally.
Official
COMIRNATY should not be mixed in the same syringe with any other vaccines or medicinal
products.
For precautions to be taken before administering COMIRNATY, see Section 4.4 Special
warnings and precautions for use.
Information
COMIRNATY (orange cap, must dilute)
Vials have an orange cap and after
dilution contain ten doses of 0.2 mL of vaccine. In order
to extract ten doses from a single vial, low dead-volume syringes and/or needles should be
used. The low dead-volume syringe and needle combination should have a dead volume of no
more than 35 microlitres. If standard syringes and needles are used, there may not be sufficient
volume to extract a tenth dose from a single vial. Irrespective of the type of syringe and needle:
• Each dose must contain 0.2 mL of vaccine.
Act
• If the amount of vaccine remaining in the vial cannot provide a full dose of 0.2 mL, discard
the vial and any excess volume.
1982
• Do not pool excess vaccine from multiple vials.
For instructions on thawing, handling, dilution and dose preparation of COMIRNATY (orange
cap, must dilute) see Section 6.6 Special precautions for disposal and other handling.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of
excipients.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 2 of 32
4.4 Special warnings and precautions for use
Traceability
In order to improve the traceability of biological medicinal products, the name and the batch
number of the administered product should be clearly recorded.
General recommendations
Released
Hypersensitivity and anaphylaxis
Events of anaphylaxis have been reported. Appropriate medical treatment and supervision
should always be readily available in case of an anaphylactic reaction following the
administration of COMIRNATY.
The individual should be kept under close observation for at least 15 minutes following
vaccination. A second dose of COMIRNATY should not be given to those who have
under
experienced anaphylaxis to the first dose of COMIRNATY.
Myocarditis and pericarditis
Very rare cases of myocarditis and pericarditis have been observed following vaccination with
the
COMIRNATY. These cases have primarily occurred within 14 days following vaccination,
more often after the second vaccination, and more often, but not exclusively in younger men.
There have been reports in females. Based on accumulating data, the reporting rates of
Official
myocarditis and pericarditis after primary series in children ages 5 through <12 years are lower
than in ages 12 through 17 years. Rates of myocarditis and pericarditis in booster doses do not
appear to be higher than after the second dose in the primary series. The cases are generally
mild and individuals tend to recover within a short time following standard treatment and rest.
Cases of myocarditis and pericarditis following vaccination have rarely been associated with
severe outcomes including death.
Information
Healthcare professionals should be alert to the signs and symptoms of myocarditis and
pericarditis, including atypical presentations. Vaccinees should be instructed to seek
immediate medical attention if they develop symptoms indicative of myocarditis or pericarditis
such as (acute and persisting) chest pain, shortness of breath, or palpitations following
vaccination. Non-specific symptoms of myocarditis and pericarditis also include fatigue,
nausea and vomiting, abdominal pain, dizziness or syncope, oedema and cough. Healthcare
professionals should consult guidance and/or specialists to diagnose and treat this condition.
Act
Stress-related responses
Some individuals may have stress-related responses associated with the process of vaccination
itself. Stress-related responses are temporary and resolve on their own. They may include
1982
dizziness, fainting, palpitations, increases in heart rate, alterations in blood pressure, feeling
short of breath, tingling sensations, sweating and/or anxiety. Individuals should be advised to
bring symptoms to the attention of the vaccination provider for evaluation and precautions
should be in place to avoid injury from fainting.
Concurrent il ness
Vaccination should be postponed in individuals suffering from acute severe febrile illness or
acute infection. The presence of a minor infection and/or low grade fever should not delay
vaccination.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 3 of 32
Thrombocytopenia and coagulation disorders
As with other intramuscular injections, COMIRNATY should be given with caution in
individuals receiving anticoagulant ther apy or those with thrombocytopenia or any coagulation
disorder (such as haemophilia) because bleeding or bruising may occur following an
intramuscular administration in these individuals.
Immunocompromised individuals
Released
The efficacy, safety and immunogenicity of COMIRNATY has not been assessed in
immunocompromised individuals, including those receiving immunosuppressant therapy. The
efficacy of COMIRNATY may be lower in immunosuppressed individuals.
Duration of protection
The duration of protection afforded by COMIRNATY is unknown as it is still being determined
by ongoing clinical trials.
under
Limitations of vaccine effectiveness
As with any vaccine, vaccination with COMIRNATY may not protect all vaccine recipients.
Individuals may not be fully protected until 7 days after their second dose of COMIRNATY.
the
Use in the elderly
Clinical studies of COMIRNATY include participants 65 years of age and older and their data
Official
contributes to the overall assessment of safety and efficacy. See Section 5.1 Pharmacodynamic
properties, Clinical trials, Efficacy against COVID-19.
Paediatric use
The safety and efficacy of COMIRNATY in children aged less than 5 years of age have not
Information
yet been established.
Effects on laboratory tests
No data available.
4.5 Interactions with other medicines and other forms of interactions
No interaction studies have been performed.
Act
Concomitant administration of COMIRNATY with other vaccines has not been studied.
1982
4.6 Fertility, pregnancy and lactation
Fertility
In a combined fertility and developmental toxicity study, female rats were intramuscularly
administered COMIRNATY prior to mating and during gestation (4 full human doses of
30 micrograms each, spanning between pre-mating day 21 and gestation day 20). SARS-CoV-
2 neutralising antibodies were present in maternal animals from prior to mating to the end of
the study on postnatal day 21 as well as in fetuses and offspring. There were no vaccine related
effects on female fertility and pregnancy rate.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 4 of 32
Pregnancy
There is limited experience with use of COMIRNATY in pregnant women. Animal studies do
not indicate direct or indirect harmful effects with respect to pregnancy, embryo/fetal
development, parturition or post-natal development (see Section 4.6 Fertility, pregnancy and
lactation, Fertility). Administration of COMIRNATY in pregnancy should only be considered
when the potential benefits outweigh any potential risks for the mother and fetus.
Released
Lactation
It is unknown whether tozinameran is excreted in human milk. A combined fertility and
developmental toxicity study in rats did not show harmful effects on offspring development
before weaning (see Section 4.6 Fertility, pregnancy and lactation, Fertility).
4.7 Effects on ability to drive and use machines
under
COMIRNATY has no, or negligible, influence on the ability to drive and use machines.
However, some of the effects mentioned under Section 4.8 Undesirable effects may
temporarily affect the ability to drive or use machines.
the
4.8 Undesirable effects
Summary of safety profile
Official
The safety of COMIRNATY was evaluated in participants 5 years of age and older in 3 clinical
studies that included 24,675 participants (comprised of 22,026 participants 16 years of age and
older, 1,131 adolescents 12 to 15 years of age and 1,518 children 5 to 11 years of age) that have
received at least one dose of COMIRNATY.
Additionally, 306 existing Phase 3 participants at 18 to 55 years of age received
Information a booster dose
of COMIRNATY approximately 6 months after the second dose in the non-placebo-controlled
booster dose portion of Study C4591001. The overall safety profile for the booster dose was
similar to that seen after 2 doses.
In Study C4591031, a placebo-controlled booster study, 5,081 participants 16 years of age and
older were recruited from Study C4591001 to receive a booster dose of COMIRNATY at least
6 months after the second dose. The overall safety profile for the booster dose was similar to
that seen after 2 doses.
Act
In a subset of C4591007 Phase 2/3 participants, 401 participants 5 to 11 years of age received
a booster dose of COMIRNATY at least 5 months after completing the primary series. The
overall safety profile for the booster dose was similar to that seen after the primary series.
1982
Participants 16 years of age and older – after 2 doses
In Study C4591001, a total of 22,026 participants 16 years of age or older received at least 1
dose of COMIRNATY 30 micrograms and a total of 22,021 participants 16 years of age or
older received placebo (including 138 and 145 adolescents 16 and 17 years of age in the
COMIRNATY and placebo groups, respectively). A total of 20,519 participants 16 years of
age or older received 2 doses of COMIRNATY.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 5 of 32
At the time of the analysis of Study C4591001 with a data cut-off of 13 March 2021 for the
placebo-controlled blinded follow-up period up to the participants’ unblinding dates, a total of
25,651 (58.2%) participants (13,031 COMIRNATY and 12,620 placebo) 16 years of age and
older were followed up for ≥4 months after the second dose. This included a total of 15,111
(7,704 COMIRNATY and 7,407 placebo) participants 16 to 55 years of age and a total of
10,540 (5,327 COMIRNATY and 5,213 placebo) participants 56 years and older.
Released
The most frequent adverse reactions in participants 16 years of age and older that received 2
doses were injection site pain (>80%), fatigue (>60%), headache (>50%), myalgia (>40%),
chills (>30%), arthralgia (>20%), pyrexia and injection site swelling (>10%) and were usually
mild or moderate in intensity and resolved within a few days after vaccination. A slightly lower
frequency of reactogenicity events was associated with greater age.
The safety profile in 545 subjects receiving COMIRNATY, that were seropositive for SARS-
CoV-2 at baseline, was similar to that seen in the general population.
under
Study C4591001 also included 200 participants with confirmed stable human
immunodeficiency virus (HIV) infection. The safety profile of the participants receiving
COMIRNATY (n=100) in the individuals with stable HIV infection was similar to that seen in
the general population.
the
Adolescents 12 to 15 years of age – after 2 doses
In an analysis of long term safety follow-up in S
Official tudy C4591001, 2,260 adolescents
(1,131 COMIRNATY 30 micrograms; 1,129 placebo) were 12 to 15 years of age. Of these,
1,559 adolescents (786 COMIRNATY and 773 placebo) have been followed for ≥ 4 months
after the second dose of COMIRNATY. The safety evaluation in Study C4591001 is ongoing.
The most frequent adverse reactions in adolescents 12 to 15 years of age that received 2 doses
Information
were injection site pain (>90%), fatigue and headache (>70%), myalgia and chills (>40%),
arthralgia and pyrexia (>20%).
Children 5 to 11 years of age – after 2 doses
In an analysis of Study C4591007 Phase 2/3, 2,268 children (1,518 COMIRNATY
10 micrograms; 750 placebo) were 5 to 11 years of age. Of these, 2,158 (95.1%)
(1,444 COMIRNATY 10 micrograms and 714 placebo) children have been followed for at
least 2 months after the second dose. The safety evaluation in Study C4591007 is ongoing.
Act
The most frequent adverse reactions in children 5 to 11 years of age that received 2 doses
included injection site pain (>80%), fatigue (>50%), headache (>30%), injection site redness
and swelling (>20%), myalgia and chills (>10%).
1982
Participants 16 years of age and older – after booster dose
A subset from Study C4591001 Phase 2/3 participants of 306 adults 18 to 55 years of age who
completed the original COMIRNATY 2-dose course, received a booster dose of
COMIRNATY approximately 6 months (range of 4.8 to 8.0 months) after receiving Dose 2.
Of these, 301 participants have been followed for ≥4 months after the booster dose of
COMIRNATY.
The most frequent adverse reactions in participants 18 to 55 years of age were injection site
pain (>80%), fatigue (>60%), headache (>40%), myalgia (>30%), chills and arthralgia (>20%).
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 6 of 32
In Study C4591031, a placebo-controlled booster study, participants 16 years of age and older
recruited from Study C4591001 received a booster dose of COMIRNATY (5,081 participants),
or placebo (5,044 participants) at least 6 months after the second dose of COMIRNATY.
Overall, participants who received a booster dose, had a median follow-up time of 2.8 months
(range 0.3 to 7.5 months) after the booster dose in the blinded placebo-controlled follow-up
period to the cut-off date (8 February 2022). Of these, 1281 participants (895 COMIRNATY
and 386 placebo) have been followed for ≥ 4 months after the booster dose of COMIRNATY.
Released
Children 5 to 11 years of age – after booster dose
In a subset from C4591007, a total of 401 children 5 to 11 years of age received a booster dose
of COMIRNATY 10 mcg at least 5 months (range of 5 to 9 months) after completing the
primary series. The analysis of the C4591007 Phase 2/3 subset is based on data up to the cut-
off date of 22 March 2022 (median follow-up time of 1.3 months).
The most frequent adverse reactions in participants 5 to 11 years of age were injection site pain
under
(>70%), fatigue (>40%), headache (>30%), myalgia, chills, injection site redness, and swelling
(>10%).
Tabulated list of adverse reactions from clinical studies and post-authorisation
experience
the
Adverse reactions observed during clinical studies are listed below according to the following
frequency categories:
Official
Very common (≥ 1/10),
Common (≥ 1/100 to < 1/10),
Uncommon (≥ 1/1,000 to < 1/100),
Rare (≥ 1/10,000 to < 1/1,000),
Information
Very rare (< 1/10,000),
Not known (cannot be estimated from the available data).
Table 1: Adverse reactions from COMIRNATY clinical trials: Individuals 12 years of age
and older
Rare
Not known
System Organ
Very
Common
Uncommon
(≥ 1/10,000
(cannot be
Act
Class
common (≥ 1/100 to
(≥ 1/1,000 to
estimated from
(≥ 1/10)
< 1/10)
< 1/100)
to
< 1/1,000)
the available
data)
Blood and
Lymphadenopathya
1982
lymphatic
system disorders
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 7 of 32
Rare
Not known
System Organ
Very
Common
Uncommon
(≥ 1/10,000
(cannot be
Class
common (≥ 1/100 to
(≥ 1/1,000 to
estimated from
(≥ 1/10)
< 1/10)
< 1/100)
to
< 1/1,000)
the available
data)
Metabolism and
Decreased appetite
nutrition
disorders
Released
Psychiatric
Insomnia
disorders
Nervous system Headache
Lethargy
Acute
disorders
peripheral
facial
paralysisb
Gastrointestinal
Nausea;
disorders
under
Skin and
Hyperhidrosis;
subcutaneous
Night sweats
tissue disorders
Musculoskeletal Arthralgia;
and connective
Myalgia the
tissue disorders
General
Injection
Injection
Asthenia; Malaise;
Facial swellingd
disorders and
site pain;
site redness
Official
administration
Fatigue;
site conditions
Chills;
Pyrexiac;
Injection
site
swelling
Information
a A higher frequency of lymphadenopathy (2.8% vs 0.4%) was observed in participants receiving a booster dose
in Study C4591031 compared to participants receiving 2 doses.
b Through the clinical trial safety follow-up period to 14 November 2020, acute peripheral facial paralysis (or
palsy) was reported by four participants in the COMIRNATY group. Onset was Day 37 after Dose 1 (participant
did not receive Dose 2) and Days 3, 9, and 48 after Dose 2. No cases of acute peripheral facial paralysis (or palsy)
were reported in the placebo group.
c A higher frequency of pyrexia was observed after the second dose compared to the first dose. The preferred term
pyrexia is a cluster term covering also body temperature increased.
d Facial swelling in vaccine recipients with a history of injection of dermatological fillers
Act
Table 2. Adverse Reactions from COMIRNATY clinical trial: Individuals 5 to 11
Years of Age (06 September 2021 Data Cut-off Date)
Rare
1982
Very
Common Uncommon ≥1/10,000
Not known
System Organ
Common ≥1/100 to ≥1/1,000 to
to
Very Rare (cannot be
Class
≥1/10
<1/10
<1/100
<1/1,000 <1/10,000 estimated
(≥10%)
(≥1% to (≥0.1% to
(<0.01%)
from the
<10%)
<1%)
(≥0.01% to
<0.1%)
available data)
Blood and
Lymphaden
lymphatic system
opathya
disorders
Immune system
Urticariab,c;
Anaphylaxisb
disorders
Pruritusb,c;
Rashb,c
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 8 of 32
Table 2. Adverse Reactions from COMIRNATY clinical trial: Individuals 5 to 11
Years of Age (06 September 2021 Data Cut-off Date)
Rare
Very
Common Uncommon ≥1/10,000
Not known
System Organ
Common ≥1/100 to ≥1/1,000 to
to
Very Rare (cannot be
Class
≥1/10
<1/10
<1/100
<1/1,000 <1/10,000 estimated
(≥10%)
(≥1% to (≥0.1% to
(<0.01%)
from the
<10%)
<1%)
(≥0.01% to
<0.1%)
available data)
Released
Metabolism and
Decreased
nutrition disorders
appetite
Nervous system
Headache
disorders
Gastrointestinal
Diarrhoeab; Nausea
disorders
Vomitingb
Musculoskeletal
Myalgia
Arthralgia Pain in
and connective
extremity
under
tissue disorders
(arm)b
General disorders Injection Pyrexia
Malaise
and administration site pain;
site conditions
Fatigue;
Chills; the
Injection
site
swelling;
Official
Injection
site redness
a. A higher frequency of lymphadenopathy was observed in C4591007 (2.5% vs. 0.9%) in
participants receiving a booster dose compared to participants receiving 2 doses.
b. These adverse reactions were identified in the post-authorisation period. The following events
were not reported in participants 5 to 11 Years of Age in Study C4591007 but were reported in
Information
individuals ≥16 years of age in Study C4591001: angioedema, lethargy, asthenia, hyperhidrosis,
and night sweats.
c. The following events are categorised as hypersensitivity reactions: urticaria, pruritus, and rash
Post-marketing experience
Although the events listed in Table 3 were not observed in the clinical trials, they are considered
adverse drug reactions for COMIRNATY as they were reported in the post-marketing
experience. As these reactions were derived from spontaneous reports, the frequencies could
not be determined and are thus considered as not known.
Act
Table 3: Adverse reactions from COMIRNATY post marketing experience
System Organ Class
Adverse Drug Reaction
1982
Immune system disorders
Anaphylaxis
Hypersensitivity reactions (e.g. rash, pruritis, urticaria, angioedema)
Cardiac disorders
Myocarditis
Pericarditis
Nervous system disorders
Dizziness
Gastrointestinal disorders
Diarrhoea
Vomiting
Musculoskeletal and connective Pain in extremity (arm)a
tissue disorders
General disorders and
Extensive swelling of vaccinated limb
administration site conditions
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 9 of 32
System Organ Class
Adverse Drug Reaction
a A higher frequency of pain in extremity (1.1% vs. 0.8%) was observed in participants receiving a booster dose in
Study C4591031 compared to participants receiving 2 doses.
Reporting suspected adverse effects
Reporting suspected adverse reactions after authorisation of the medicine is important. It allows
continued monitoring of the benefit/risk balance of the medicine. Healthcare professionals are
Released
asked to report any suspected adverse reactions at
https://nzphvc.otago.ac.nz/reporting/.
4.9 Overdose
Overdose data is available from 52 study participants included in the clinical trial that due to
an error in dilution received 58 micrograms of COMIRNATY. The COMIRNATY recipients
did not report an increase in reactogenicity or adverse reactions.
under
In the event of overdose, monitoring of vital functions and possible symptomatic treatment is
recommended.
For advice on the management of overdose please contact the National Poisons Centre on 0800
POISON (0800 764766). the
5. PHARMACOLOGICAL PROPERTIES
Official
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: vaccines, other viral vaccines, ATC code: J07BX03.
Information
Mechanism of action
The nucleoside-modified messenger RNA in COMIRNATY is formulated in lipid
nanoparticles, which enable delivery of the non-replicating RNA into host cells to direct
transient expression of the SARS-CoV-2 spike (S) antigen. The mRNA codes for membrane-
anchored, full-length S with two point mutations within the central helix. Mutation of these
two amino acids to proline locks S in an antigenically preferred prefusion conformation.
COMIRNATY elicits both neutralising antibody and cellular immune responses to the antigen,
which may contribute to protection against COVID-19.
Act
Clinical efficacy and safety
Efficacy
1982
Study C4591001 is a multicentre, multinational, Phase 1/2/3 randomised, placebo-controlled,
observer-blind dose-finding, vaccine candidate selection and efficacy study in participants 12
years of age and older. Randomisation was stratified by age: 12 to 15 years of age, 16 to 55
years of age, or 56 years of age and older, with a minimum of 40% of participants in the ≥56-
year stratum. The study excluded participants who were immunocompromised and those who
had previous clinical or microbiological diagnosis of COVID-19. Participants with pre-existing
stable disease, defined as disease not requiring significant change in therapy or hospitalisation
for worsening disease during the 6 weeks before enrolment, were included as were participants
with known stable infection with HIV, hepatitis C virus (HCV) or hepatitis B virus (HBV).
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 10 of 32
Efficacy in participants 16 years of age and older – after 2 doses
In the Phase 2/3 portion of Study C4591001, based on data accrued through
14 November 2020, approximately 44,000 participants were randomised equally and were to
receive 2 doses of COMIRNATY or placebo. The efficacy analyses included participants that
received their second vaccination within 19 to 42 days after their first vaccination. The majority
(93.1%) of vaccine recipients received the second dose 19 days to 23 days after Dose 1.
Participants are planned to be followed for up to 24 months after Dose 2, for assessments of
Released
safety and efficacy against COVID-19. In the clinical study, participants were required to
observe a minimum interval of 14 days before and after administration of an influenza vaccine
in order to receive either placebo or COMIRNATY. In the clinical study, participants were
required to observe a minimum interval of 60 days before or after receipt of blood/plasma
products or immunoglobulins through to conclusion of the study in order to receive either
placebo or COMIRNATY.
The population for the analys
under is of the primary efficacy endpoint included 36,621 participants
12 years of age and older (18,242 in the COMIRNATY group and 18,379 in the placebo group)
who did not have evidence of prior infection with SARS-CoV-2 through 7 days after the second
dose. In addition, 134 participants were between the ages of 16 to 17 years of age (66 in the
COMIRNATY group and 68 in the placebo group) and 1616 participants 75 years of age and
the
older (804 in the COMIRNATY group and 812 in the placebo group).
At the time of the primary efficacy analysis, participants had been followed for symptomatic
Official
COVID-19 for in total 2,214 person-years for the COMIRNATY group and in total 2,222
person-years for the placebo group.
There were no meaningful clinical differences in overall vaccine efficacy in participants who
were at risk of severe COVID-19 including those with 1 or more comorbidities that increase
the risk of severe COVID-19 (e.g. asthma, body mass index (BMI) ≥30 kg/m2, chronic
Information
pulmonary disease, diabetes mellitus, hypertension).
COMIRNATY efficacy information is presented in Table 4.
Table 4: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2, by age
subgroup – participants without evidence of infection prior to 7 days after Dose 2 –
evaluable efficacy (7 days) population
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
of prior SARS-CoV-2 infection*
Act
COMIRNATY
Placebo
Na = 18,198 Cases
Na = 18,325 Cases
Vaccine efficacy
Subgroup
n1b
n1b
% 1982
Surveillance timec
Surveillance timec
(95% CI)f
(n2d)
(n2d)
All participantse
8
162
95.0
2.214 (17,411)
2.222 (17,511)
(90.0, 97.9)
16 to 64 years
7
143
95.1
1.706 (13,549)
1.710 (13,618)
(89.6, 98.1)
65 years and older
1
19
94.7
0.508 (3848)
0.511 (3880)
(66.7, 99.9)
65 to 74 years
1
14
92.9
0.406 (3074)
0.406 (3095)
(53.1, 99.8)
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 11 of 32
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Na = 18,198 Cases
Na = 18,325 Cases
Vaccine efficacy
Subgroup
n1b
n1b
%
Surveillance timec
Surveillance timec
(95% CI)f
(n2d)
(n2d)
Released
75 years and older
0
5
100.0
0.102 (774)
0.106 (785)
(-13.1, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 [*Case definition: (at least 1 of) fever, new or increased cough,
new or increased shortness of breath, chills, new or increased muscle pain, new loss of taste or smell, sore
throat, diarrhoea or vomiting.]
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the last dose) of
past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not
detected by nucleic acid amplification tests (NAAT) [nasal swab] at Visits 1 and 2), and had negative NAAT
under
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of
the surveillance period.
the
d. n2 = Number of participants at risk for the endpoint.
e. No confirmed cases were identified in adolescents 12 to 15 years of age.
f. Two-sided confidence interval (CI) for vaccine efficacy (VE) is derived based on the Clopper and Pearson
Official
method adjusted to the surveillance time. CI not adjusted for multiplicity.
In the second primary analysis, efficacy of COMIRNATY in preventing first COVID-19
occurrence from 7 days after Dose 2 compared to placebo was 94.6% (95% credible interval
of 89.9% to 97.3%) in participants 16 years of age and older with or without evidence of prior
infection with SARS-CoV-2.
Information
Additionally, subgroup analyses of the primary efficacy endpoint showed similar efficacy point
estimates across genders, ethnic groups, and participants with medical comorbidities associated
with high risk of severe COVID-19.
Updated efficacy analyses were performed with additional confirmed COVID-19 cases accrued
during blinded placebo-controlled follow-up through 13 March 2021, representing up to
6 months of follow-up after Dose 2 for participants in the efficacy population. Act
The updated vaccine efficacy information is presented in Table 5.
1982
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 12 of 32
Table 5: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2, by age
subgroup – participants without evidence of infection prior to 7 days after Dose 2 –
evaluable efficacy (7 days) population during the placebo-controlled follow-up period
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Na=20,998
Na=21,096
Released
Cases
Cases
n1b
n1b
Surveil ance Timec
Surveil ance Timec
Vaccine efficacy %
Subgroup
(n2d)
(n2d)
(95% CIe)
All participantsf
77
850
91.3
6.247 (20,712)
6.003 (20,713)
(89.0, 93.2)
16 to 64 years
70
710
90.6
4.859 (15,519)
4.654 (15,515)
(87.9, 92.7)
under
65 years and older
7
124
94.5
1.233 (4192)
1.202 (4226)
(88.3, 97.8)
65 to 74 years
6
98
94.1
0.994 (3350)
0.966 (3379)
(86.6, 97.9)
75 years and older
1
26
96.2
the
0.239 (842)
0.237 (847)
(76.9, 99.9)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
Official
diarrhoea; vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative
at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
Information
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Two-sided confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method
adjusted to the surveillance time.
f. Included confirmed cases in participants 12 to 15 years of age: 0 in the COMIRNATY group (both without
and with or without evidence of prior SARS-CoV-2 infection); 16 and 18 in the placebo group (without and
with or without evidence of prior SARS-CoV-2 infection, respectively).
Act
Efficacy against severe COVID-19 in participants 12 years of age or older – after 2 doses
As of 13 March 2021, vaccine efficacy against severe COVID-19 is presented only for
participants with or without prior SARS-CoV-2 infection (Table 6) as the COVID-19 case
1982
counts in participants without prior SARS-CoV-2 infection were the same as those in
participants with or without prior SARS-CoV-2 infection in both the COMIRNATY and
placebo groups.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 13 of 32
Table 6. Vaccine Efficacy – First Severe COVID-19 Occurrence in Participants With
or Without* Prior SARS-CoV-2 Infection Based on Food and Drug Administration
(FDA)† Definition After Dose 1 or From 7 Days After Dose 2 in the Placebo-Controlled
Follow-up
COMIRNATY
Placebo
Cases
Cases
Released
n1a
n1a
Vaccine Efficacy %
Surveil ance Time (n2b) Surveil ance Time (n2b)
(95% CIc)
1
30
96.7
After Dose 1d
8.439e (22,505)
8.288e (22,435)
(80.3, 99.9)
1
21
95.3
7 days after Dose 2f
6.522g (21,649)
6.404g (21,730)
(70.9, 99.9)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at
least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or increased
shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat; diarrhoea;
under
vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative
at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
† Severe illness from COVID-19 as defined by FDA is confirmed COVID-19 and presence of at least 1 of the
following:
the
• Clinical signs at rest indicative of severe systemic illness (respiratory rate ≥30 breaths per minute, heart
rate ≥125 beats per minute, saturation of oxygen ≤93% on room air at sea level, or ratio of arterial
oxygen partial pressure to fractional inspired oxygen <300 mm Hg);
Official
• Respiratory failure [defined as needing high-flow oxygen, noninvasive ventilation, mechanical
ventilation or extracorporeal membrane oxygenation (ECMO)];
• Evidence of shock (systolic blood pressure <90 mm Hg, diastolic blood pressure <60 mm Hg, or
requiring vasopressors);
• Significant acute renal, hepatic, or neurologic dysfunction;
• Admission to an Intensive Care Unit;
Information
• Death.
a. n1 = Number of participants meeting the endpoint definition.
b. n2 = Number of participants at risk for the endpoint.
c. Two-side confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method
adjusted to the surveillance time.
d. Efficacy assessed based on the Dose 1 all available efficacy (modified intention-to-treat) population that
included all randomised participants who received at least 1 dose of study intervention.
e. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from Dose 1 to the end of the surveillance
period.
f. Efficacy assessed based on the evaluable efficacy (7 Days) population that included al eligible randomised
Act
participants who receive all dose(s) of study intervention as randomised within the predefined window, have
no other important protocol deviations as determined by the clinician
g. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
1982
surveillance period.
Efficacy and immunogenicity in adolescents 12 to 15 years of age – after 2 doses
An analysis of Study C4591001 has been performed in adolescents 12 to 15 years of age up to
a data cutoff date of 13 March 2021.
The vaccine efficacy information in adolescents 12 to 15 years of age is presented in Table 7.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 14 of 32
Table 7: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2 –
participants without evidence of infection and with or without evidence of infection prior
to 7 days after Dose 2 – adolescents 12 to 15 years of age evaluable efficacy (7 days)
population
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
without evidence of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Released
Na = 1005
Na = 978
Cases
Cases
n1b
n1b
Vaccine efficacy
Surveil ance timec (n2d) Surveil ance timec (n2d)
% (95% CIe)
Adolescents
0
16
12 to 15 years
0.154 (1001)
0.147 (972)
100.0 (75.3, 100.0)
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
with or without* evidence of prior SARS-CoV-2 infection
under
COMIRNATY
Placebo
Na = 1119
Na = 1110
Cases
Cases
n1b
n1b
Vaccine efficacy
Surveil ance timec (n2d) Surveil ance timec (n2d)
% (95% CIe)
the
Adolescents
0
18
12 to 15 years
0.170 (1109)
0.163 (1094)
100.0 (78.1, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at
Official
least 1 symptom consistent with COVID-19 [*Case definition: (at least 1 of) fever, new or increased cough, new
or increased shortness of breath, chills, new or increased muscle pain, new loss of taste or smell, sore throat,
diarrhoea or vomiting).
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the last dose) of
past SARS-CoV-2 infection (i.e, N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not
detected by nucleic acid amplification tests (NAAT) [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
Information
a. N = number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all subjects within each group at
risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of subjects at risk for the endpoint.
e. Confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method adjusted
for surveillance time. CI not adjusted for multiplicity.
In Study C4591001 an analysis of SARS-CoV-2 neutralising titres in a randomly selected
Act
subset of participants was performed to demonstrate non-inferior immune responses (within
1.5-fold) comparing adolescents 12 to 15 years of age to participants 16 to 25 years of age who
had no serological or virological evidence of past SARS-CoV-2 infection. The immune
1982
response to COMIRNATY in adolescents 12 to 15 years of age (n = 190) was non-inferior to
the immune response in participants 16 to 25 years of age (n = 170), based on results for SARS-
CoV-2 neutralising titres at 1 month after Dose 2. The geometric mean titres (GMT) ratio of
the adolescents 12 to 15 years of age group to the participants 16 to 25 years of age group was
1.76, with a 2-sided 95% CI of 1.47 to 2.10, meeting the 1.5-fold non-inferiority criterion (the
lower bound of the 2-sided 95% CI for the geometric mean ratio [GMR] >0.67), which
indicates a statistically greater response in the adolescents 12 to 15 years of age than that of
participants 16 to 25 years of age.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 15 of 32
An updated efficacy analysis of Study C4591001 has been performed in approximately 2,260
adolescents 12 to 15 years of age evaluating confirmed COVID-19 cases accrued up to a data
cut-off date of 2 September 2021, representing up to 6 months of follow-up after Dose 2 for
participants in the efficacy population.
The updated vaccine efficacy information in adolescents 12 to 15 years of age is presented in
Table 8.
Released
Table 8: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Dose 2:
Without Evidence of Infection and With or Without Evidence of Infection
Prior to 7 Days After Dose 2 – Blinded Placebo-Controlled Follow-up Period,
Adolescents 12 To 15 Years of Age Evaluable Efficacy (7 Days) Population
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
without evidence of prior SARS-CoV-2 infection*
under
COMIRNATY
Placebo
Na=1057
Na=1030
Cases
Cases
n1b
n1b
Surveil ance Timec
Surveil ance Timec
Vaccine Efficacy %
(n2d)
(n2d)
(95% CIe)
the
Adolescents
0
28
100.0
12 to 15 years of age
0.343 (1043)
0.322 (1019)
(86.8, 100.0)
First COVID-19 occurrence from 7 days after D
Official
ose 2 in adolescents 12 to 15 years of
age with or without evidence of prior SARS-CoV-2 infection
COMIRNATY
Placebo
Na=1119
Na=1109
Cases
Cases
n1b
n1b
Surveil ance Timec
Surveil ance Timec
Vaccine Efficacy %
Information
(n2d)
(n2d)
(95% CIe)
Adolescents
0
30
100.0
12 to 15 years of age
0.362 (1098)
0.345 (1088)
(87.5, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum]
negative at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had
negative NAAT (nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the
Act
analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
1982
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each
group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the
end of the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Two-sided confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson
method adjusted for surveillance time.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 16 of 32
Efficacy in children 5 to 11 years of age – after 2 doses
A descriptive efficacy analysis of Study C4591007 has been performed in 1,968 children 5 to
11 years of age without evidence of infection prior to 7 days after Dose 2. This analysis
evaluated confirmed symptomatic COVID-19 cases accrued up to a data cut-off date of 8
October 2021.
The descriptive vaccine efficacy results in children 5 to 11 years of age without evidence of
Released
prior SARS-CoV-2 infection are presented in Table 9. None of the cases accrued met criteria
for severe COVID-19 or multisystem inflammatory syndrome in children (MIS-C). No cases
of COVID-19 were observed in either the vaccine group or the placebo group in participants
with evidence of prior SARS-CoV-2 infection.
Table 9: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Dose 2:
Without Evidence of Infection Prior to 7 Days After Dose 2 – Phase 2/3 – Children 5 To
11 Years of Age Evaluable Efficacy Population
under
First COVID-19 occurrence from 7 days after Dose 2 in children 5 to 11 years of age
without evidence of prior SARS-CoV-2 infection*
COMIRNATY±
10 micrograms/dose
Placebo
the
Na=1305
Na=663
Cases
Cases
n1b
n1b
Official
Surveil ance Timec
Surveil ance Timec
Vaccine Efficacy %
(n2d)
(n2d)
(95% CI)
Children 5 to
3
16
90.7
11 years of age
0.322 (1273)
0.159 (637)
(67.7, 98.3)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
Information
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum]
negative at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had
negative NAAT (nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the
analysis.
± Pfizer-BioNTech COVID-19 Vaccine (10 micrograms modRNA).
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each
group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the
Act
end of the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
1982
Immunogenicity in children 5 to 11 years of age – after 2 doses
Study C4591007 is a Phase 1/2/3 study comprised of an open-label vaccine dose-finding
portion (Phase 1) and a multicentre, multinational, randomised, saline placebo-controlled,
observer-blind efficacy portion (Phase 2/3) that has enrolled participants 5 to 11 years of age.
In C4591007, an analysis of SARS-CoV-2 50% neutralising titres (NT50) 1 month after Dose
2 in a randomly selected subset of participants demonstrated effectiveness by immunobridging
of immune responses comparing children 5 to 11 years of age in the Phase 2/3 part of Study
C4591007 to participants 16 to 25 years of age in the Phase 2/3 part of Study C4591001 who
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 17 of 32
had no serological or virological evidence of past SARS-CoV-2 infection up to 1 month after
Dose 2, meeting the prespecified immunobridging criteria for both the geometric mean ratio
(GMR) and the seroresponse difference with seroresponse defined as achieving at least 4-fold
rise in SARS-CoV-2 NT50 from baseline (before Dose 1).
The ratio of the SARS-CoV-2 NT50 in children 5 to 11 years of age to that of young adults 16
to 25 years of age was 1.04 (2-sided 95% CI: 0.93, 1.18), as presented in Table 10.
Released
Table 10: Summary of geometric mean ratio for 50% neutralising titre – Comparison
of children 5 to 11 years of age (Study C4591007) to participants 16 to 25 years of age
(Study C4591001) – participants without* evidence of infection up to 1 month after Dose
2 – evaluable immunogenicity population
COMIRNATY
5 to 11 years/
10 microgram/dose 30 microgram/dose
16 to 25 years
5 to 11 years
16 to 25 years
under
na=264
na=253
Met
Assay
Time
GMTc
GMTc
GMRd immunobridging
pointb
(95% CIc)
(95% CIc)
(95% CId)
objectivee
(Y/N)
the
SARS-CoV-2
neutralisation 1 month
1197.6
1146.5
1.04
assay - NT50 after
(1106.1, 1296.6)
(1045.5, 1257.2) (0.93, 1.18)
Y
Official
(titre)f
Dose 2
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre;
LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test; NT50 = 50% neutralising titre;
SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
*Participants who had no serological or virological evidence (up to 1 month post-Dose 2 blood sample collection)
of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and 1 month after Dose
2, SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2, and negative NAAT (nasal swab) at
Information
any unscheduled visit up to 1 month after Dose 2 blood collection) and had no medical history of COVID-
19 were included in the analysis.
a. n = Number of participants with valid and determinate assay results for the specified assay at the given
dose/sampling time point.
b. Protocol-specified timing for blood sample collection.
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
d. GMRs and 2-sided 95% CIs were calculated by exponentiating the mean difference of the logarithms of the
titres (Group 1[5 to 11 years of age] - Group 2 [16 to 25 years of age]) and the corresponding CI (based on
Act
the Student t distribution).
e. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67 and
the point estimate of the GMR is ≥0.8.
f. SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation
1982
Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus
neutralisation is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution
at which 50% of the virus is neutralised.
Among participants without prior evidence of SARS-CoV-2 infection up to 1 month after
Dose 2, 99.2% of children 5 to 11 years of age and 99.2% of participants 16 to 25 years of age
had a seroresponse from before vaccination to 1 month after Dose 2. The difference in
proportions of participants who had seroresponse between the 2 age groups (children – young
adult) was 0.0% (2-sided 95% CI: -2.0%, 2.2%) as presented in Table 11.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 18 of 32
Table 11: Difference in percentages of participants with seroresponse – participants
without evidence of infection up to 1 month after Dose 2 – immunobridging subset –
Phase 2/3 – comparison of 5 to 11 years of age to Study C4591001 Phase 2/3 16 to 25 years
of age – evaluable immunogenicity population
COMIRNATY
10
30
5 to 11 years/
microgram/dose microgram/dose
16 to 25 years
Released
5 to 11 years
16 to 25 years
Na=264
Na=253
Met
Assay
Time
nc (%)
nc (%)
Difference %e immunobridging
pointb
(95% CId)
(95% CId)
(95% CIf)
objectiveg
(Y/N)
SARS-CoV-2
neutralisation
1 month
262 (99.2)
251 (99.2)
0.0
under
assay – NT50 after
(97.3, 99.9)
(97.2, 99.9)
(-2.0, 2.2)
Y
(titre)h
Dose 2
Abbreviations: LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test;
N-binding = SARS-CoV-2 nucleoprotein–binding; NT50 = 50% neutralising titre 50; SARS-CoV-2 = severe acute
respiratory syndrome coronavirus 2.
Note: Seroresponse is defined as achieving a ≥4-fold rise from baseline (before Dose 1). If the baseline measurement
the
is below the LLOQ, a postvaccination assay result ≥4 × LLOQ is considered a seroresponse.
Note: Participants who had no serological or virological evidence (up to 1 month post-Dose 2 blood sample
collection) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and 1 month after
Official
Dose 2, SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2, and negative NAAT (nasal swab) at
any unscheduled visit up to 1 month after Dose 2 blood collection) and had no medical history of COVID-19 were
included in the analysis.
a. N = number of participants with valid and determinate assay results both before vaccination and at 1 month after
Dose 2. These values are the denominators for the percentage calculations.
b. Protocol-specified timing for blood sample collection.
c. n = Number of participants with seroresponse for the given assay at the given dose/sampling time point.
Information
d. Exact 2-sided CI based on the Clopper and Pearson method.
e. Difference in proportions, expressed as a percentage (Group 1 [5 to 11 years of age] – Group 2 [16 to 25 years
of age]).
f. 2-Sided CI, based on the Miettinen and Nurminen method for the difference in proportions, expressed as a
percentage.
g. Immunobridging is declared if the lower bound of the 2-sided 95% CI for the difference in proportions is greater
than -10.0%.
h. SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation Assay.
The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus neutralisation is
read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution at which 50% of
the virus is neutralised.
Act
Immunogenicity in participants 18 years of age and older – after booster dose
1982
Effectiveness of a booster dose of COMIRNATY was based on an assessment of 50%
neutralising titres (NT50) against SARS-CoV-2 (USA_WA1/2020). In Study C4591001,
analyses of NT50 1 month after the booster dose compared to 1 month after the primary series
in individuals 18 to 55 years of age who had no serological or virological evidence of past
SARS-CoV-2 infection up to 1 month after the booster vaccination demonstrated
noninferiority for both GMR and difference in seroresponse rates. Seroresponse for a
participant was defined as achieving a ≥4-fold rise in NT50 from baseline (before Dose 1),
These analyses are summarised in Table 12.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 19 of 32
Table 12. SARS-CoV-2 neutralisation assay - NT50 (titre)† (SARS-CoV-2
USA_WA1/2020) – GMT and seroresponse rate comparison of 1 month after booster dose
to 1 month after primary series – participants 18 to 55 years of age without evidence of
infection up to 1 month after booster dose* – booster dose evaluable immunogenicity
population±
1 month after
booster dose/-
Released
1 month after
Met
1 month after
1 month after
primary
noninferiority
booster dose
primary series
series
objective
n
(95% CI)
(95% CI)
(97.5% CI)
(Y/N)
Geometric mean
50% neutralising
2466.0b
755.7
b
3.26c
titre (GMTb)
212a (2202.6, 2760.8) (663.1, 861.2)
(2.76, 3.86)
Yd
Seroresponse rate
199f
190f
(%) for 50%
99.5%
95.0%
4.5%g
under
neutralising titre† 200e (97.2%, 100.0%) (91.0%, 97.6%) (1.0%, 7.9%
h)
Yi
Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titre;
LLOQ = lower limit of quantitation; N-binding = SARS-CoV-2 nucleoprotein-binding; NAAT = nucleic acid
amplification test; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute respiratory syndrome
coronavirus 2; Y/N = yes/no. the
† SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralisation
Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus
neutralisation is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum
dilution at which 50% of the virus is neutralised.
Official
* Participants who had no serological or virological evidence (up to 1 month after receipt of a booster dose
of Comirnaty) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative and
SARS-CoV-2 not detected by NAAT [nasal swab]) and had a negative NAAT (nasal swab) at any
unscheduled visit up to 1 month after the booster dose were included in the analysis.
± All eligible participants who had received 2 doses of Comirnaty as initial y randomised, with Dose 2
received within the predefined window (within 19 to 42 days after Dose 1), received a booster dose of
Information
Comirnaty, had at least 1 valid and determinate immunogenicity result after booster dose from a blood
col ection within an appropriate window (within 28 to 42 days after the booster dose), and had no other
important protocol deviations as determined by the clinician.
a. n = Number of participants with valid and determinate assay results at both sampling time points within
specified window.
b. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
c. GMRs and 2-sided 97.5% CIs were calculated by exponentiating the mean differences in the logarithms of
the assay and the corresponding CIs (based on the Student t distribution).
d. Noninferiority is declared if the lower bound of the 2-sided 97.5% CI for the GMR is > 0.67 and the point
Act
estimate of the GMR is ≥ 0.80.
e. n = Number of participants with valid and determinate assay results for the specified assay at baseline,
1 month after Dose 2 and 1 month after the booster dose within specified window. These values are the
denominators for the percentage calculations.
1982
f. Number of participants with seroresponse for the given assay at the given dose/sampling time point. Exact
2-sided CI based on the Clopper and Pearson method.
g. Difference in proportions, expressed as a percentage (1 month after booster dose – 1 month after Dose 2).
h. Adjusted Wald 2-sided CI for the difference in proportions, expressed as a percentage.
i. Noninferiority is declared if the lower bound of the 2-sided 97.5% CI for the percentage difference is
> -10%.
Relative vaccine efficacy in participants 16 years of age and older – after booster dose
An interim efficacy analysis of Study C4591031, a placebo-controlled booster study, was
performed in approximately 10,000 participants 16 years of age and older who were recruited
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 20 of 32
from Study C4591001, evaluated confirmed COVID-19 cases accrued from at least 7 days after
booster vaccination up to a data cut-off date of 8 February 2022 (a period when Delta and then
Omicron was the predominant variant), which represents a median of 2.8 months (range 0.3 to
7.5 months) post-booster follow-up. Vaccine efficacy of the COMIRNATY booster dose after
the primary series relative to the placebo booster group who only received the primary series
dose was assessed. The relative vaccine efficacy information for participants 16 years of age
and older is presented in Table 13.
Released
Table 13: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Booster
Vaccination – Participants 16 Years of Age and Older Without Evidence of
Infection and Participants With or Without Evidence of Infection Prior to 7
Days After Booster Vaccination – Evaluable Efficacy Population
First COVID-19 occurrence from 7 days after booster dose in participants without evidence
of prior SARS-CoV-2 infection*
under
COMIRNATY
Placebo
Na=4689
Na=4664
Cases
Cases
n1b
n1b
Relative Vaccine
Surveil ance Timec
Surveil ance Timec
Efficacye %
(n2d
the
)
(n2d)
(95% CIf)
First COVID-19
occurrence from
7 days after booster
63 Official
148
63.9
vaccination
1.098 (4639)
0.932 (4601)
(51.1, 73.5)
First COVID-19 occurrence from 7 days after booster dose in participants with or without
evidence of prior SARS-CoV-2 infection
COMIRNATY
Placebo
Na=4997
Na=4942
Cases
Cases
Information
n1b
n1b
Relative Vaccine
Surveil ance Timec
Surveil ance Timec
Efficacye %
(n2d)
(n2d)
(95% CIf)
First COVID-19
occurrence from
7 days after booster
67
150
62.4
vaccination
1.179 (4903)
0.989 (4846)
(49.5, 72.2)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
Act
diarrhea; vomiting).
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the booster
1982
vaccination) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and
SARS-CoV-2 not detected by NAAT [nasal swab] at Visit 1, and had a negative NAAT [nasal swab] at
any unscheduled visit prior to 7 days after booster vaccination) were included in the analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each
group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after the booster
vaccination to the end of the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Relative vaccine efficacy of the COMIRNATY booster group relative to the placebo group (non-booster).
f. Two-sided confidence interval (CI) for relative vaccine efficacy is derived based on the Clopper and
Pearson method adjusted for surveillance time.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 21 of 32
Immunogenicity in children 5 to 11 years of age – after booster dose
Effectiveness of a booster dose of COMIRNATY was based on an assessment of NT50 against
the reference strain of SARS-CoV-2 (USA_WA1/2020). Analyses of NT50 1 month after the
booster dose compared to before the booster dose demonstrated a substantial increase in GMTs
in individuals 5 to 11 years of age who had no serological or virological evidence of past
SARS-CoV-2 infection up to 1 month after the booster dose. This analysis is summarised in
Table 14.
Released
Table 14: Summary of Geometric Mean Titres – NT50 – Participants Without Evidence
of Infection – Phase 2/3 – Immunogenicity Set – 5 to 11 Years of Age –
Evaluable Immunogenicity Population
COMIRNATY 10 mcg/Dose
3-Dose Set
2-Dose Set
Total
under
Dose/
Sampling
GMTc
GMTc
GMTc
Assay
Time Pointa
nb
(95% CIc)
nb
(95% CIc) nb
(95% CIc)
20.5
20.5
20.5
1 month Prevax 79
(20.5, 20.5)
67 (20.5, 20.5) 146 (20.5, 20.5)
the
SARS-CoV-2 1 month after Dose
1659.4
1110.7
1253.9
neutralisation
2
29 (1385.1, 1988.0) 67 (965.3, 1278.1) 96 (1116.0, 1408.9)
assay - NT50
Official
271.0
271.0
(titre)
3 months Prevax 67 (229.1, 320.6) -
-
67 (229.1, 320.6)
1 month after Dose
2720.9
2720.9
3
67 (2280.1, 3247.0) -
-
67 (2280.1, 3247.0)
Abbreviations: CI = confidence interval; GMT = geometric mean titre; LLOQ = lower limit of quantitation;
NAAT = nucleic acid amplification test; N-binding = SARS-CoV-2 nucleoprotein–binding; NT50 = 50%
Information
neutralising titre; Prevax = before vaccination; SARS-CoV-2 = severe acute respiratory syndrome coronavirus
2.
Note: Three-dose immunogenicity set included the first 130 participants who received Dose 3 and completed
1-month post–Dose 3 visit prior to March 15, 2022. Among those, 30 had blood sample collection at 1-month
post-Dose 2. Two-dose immunogenicity set included an extra 67 participants randomly selected from previous
Dose-2 evaluable immunogenicity population and without evidence of infection up to 1-month post–Dose 2
subset used for 2-dose immunobridging analysis.
Note: Participants included in this analysis had no serological or virological evidence of past SARS-CoV-2
infection up to the 1-month post–Dose 2 (for 1-month post–Dose 2 time point) or 1-month post–Dose 3 (for pre–
Dose 3 and 1-month post–Dose 3 time point) study blood sample collection. Having no evidence of past SARS-
CoV-2 infection up to 1-month post–Dose 2 was defined as having a negative N-binding antibody (serum) result
Act
at the Dose 1 and 1-month post–Dose 2 study visits; a negative NAAT (nasal swab) result at the Dose 1 and
Dose 2 study visits and any unscheduled visit prior to the 1-month post–Dose 2 blood sample collection; and no
medical history of COVID-19. Having no evidence of past SARS-CoV-2 infection up to 1-month post-Dose 3
1982
was defined as having a negative N-binding antibody (serum) result at the Dose 1, 1-month post–Dose 2 (if
available), Dose 3, and 1-month post–Dose 3 study visits; a negative NAAT (nasal swab) result at the Dose 1,
Dose 2, and Dose 3 study visits and any unscheduled visit prior to the 1-month post–Dose 3 blood sample
collection; and no medical history of COVID-19.
a. Protocol-specified timing for blood sample collection.
b. n = Number of participants with valid and determinate assay results for the specified assay at the given
dose/sampling time point.
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 ×
LLOQ.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 22 of 32
Immunogenicity in children 5 to 11 years of age on the Omicron variant (B1.1.529) –
after booster dose
The neutralising GMTs against both the Omicron variant (B1.1.529) and reference strain were
substantially increased after booster vaccination compared with after the 2-dose primary series.
At 1-month post-Dose 2, the observed neutralising GMTs for the Omicron variant (B1.1.529)
and reference strain were 27.6 and 323.8, respectively. At 1-month post-Dose 3, the observed
neutralising GMTs for the Omicron variant (B1.1.529) and reference strain were 614.4 and
Released
1702.8, respectively (see Table 15).
For the Omicron variant (B1.1.529), neutralising titres after booster vaccination (1-month post-
Dose 3) increased 22-fold over those after the 2-dose primary series (1-month post-Dose 2).
For the reference strain, the increase after the booster relative to the primary series was 5.3-
fold.
Table 15: Summary of Geometric Mean Titres – Omicron-Neutralisation Subset –
under
Participants Without Evidence of Infection – Phase 2/3 – Immunogenicity Set
– 5 to 11 Years of Age – Evaluable Immunogenicity Population
COMIRNATY
the
10 mcg/Dose
Vaccine Group (as Randomised)
GMTc
Official
Assay
Time Pointb
nb
(95% CIc)
SARS-COV-2 FFRNT-
27.6
B.1.1.529 strain
1 month after Dose 2
29
(22.1, 34.5)
(Omicron) - NT50
614.4
(titre)
1 month after Dose 3
17
(410.7, 919.2)
SARS-CoV-2 FFRNT-
323.8
Information
reference strain - NT50
1 month after Dose 2
29
(267.5, 392.1)
(titre)
1702.8
1 month after Dose 3
17
(1282.6, 2260.7)
Abbreviations: CI = confidence interval; FFRNT = fluorescence focus reduction neutralisation test;
GMT = geometric mean titre; LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test;
N-binding = SARS-CoV-2 nucleoprotein–binding; NT50 = 50% neutralising titre; SARS-CoV-2 = severe acute
respiratory syndrome coronavirus 2.
Note: Participants included in this analysis had no serological or virological evidence of past SARS-CoV-2
infection up to the 1-month post–Dose 2 (for 1-month post–Dose 2 time point) or 1-month post–Dose 3 (for
1-month post–Dose 3 time point) study blood sample collection. Having no evidence of past SARS-CoV-2
infection up to 1-month post–Dose 2 was defined as having a negative N-binding antibody (serum) result at
Act
the Dose 1 and 1-month post–Dose 2 study visits; a negative NAAT (nasal swab) result at the Dose 1 and Dose
2 study visits and any unscheduled visit prior to the 1-month post–Dose 2 blood sample collection; and no
medical history of COVID-19. Having no evidence of past SARS-CoV-2 infection up to 1-month post–Dose 3
1982
was defined as having a negative N-binding antibody (serum) result at the Dose 1, 1-month post–Dose 2 (if
available), Dose 3, and 1-month post–Dose 3 study visits; a negative NAAT (nasal swab) result at the Dose 1,
Dose 2, and Dose 3 study visits and any unscheduled visit prior to the 1-month post–Dose 3 blood sample
collection; and no medical history of COVID-19.
a. Protocol-specified timing for blood sample collection.
b. n = Number of participants with valid and determinate assay results for the specified assays at the given
dose/sampling time point.
c. GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titres and the
corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to
0.5 × LLOQ.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 23 of 32
This medicine has been given a provisional consent under Section 23 of the Act. This means
that further evidence on this medicine is awaited or that there are specific conditions of use.
Refer to the consent notice published in the New Zealand Gazette for the specific conditions.
5.2 Pharmacokinetic properties
Not applicable.
Released
5.3 Preclinical safety data
Genotoxicity/Carcinogenicity
Neither genotoxicity nor carcinogenicity studies were performed. The components of
COMIRNATY (lipids and mRNA) are not expected to have genotoxic potential.
under
6. PHARMACEUTICAL PARTICULARS
6.1 List of excipients the
((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
Official
Distearoylphosphatidylcholine (DSPC)
Cholesterol
Trometamol
Trometamol hydrochloride
Information
Sucrose
Water for injections
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those
mentioned in Section 6.6 Special precautions for disposal and other handling. Act
6.3 Shelf life
1982
COMIRNATY (orange cap, must dilute)
Unopened vial
Frozen vial
24 months when stored at -90°C to -60°C.
The vaccine will be received frozen at -90°C to -60°C. Frozen vaccine can be stored either at
-90°C to -60°C or 2°C to 8°C upon receipt.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 24 of 32
When stored frozen at -90°C to -60°C, 10-vial packs of the vaccine can be thawed at 2°C to 8°C
for 4 hours or individual vials can be thawed at room temperature (up to 30°C) for 30 minutes.
Thawed vial
If the vaccine is received at 2°C to 8°C it should be stored at 2°C to 8°C. Once removed from
frozen storage, the unopened vial may be stored refrigerated at 2°C to 8°C for a single period
of up to 10 weeks within the 24-month shelf life.
Released
Upon moving the product to 2°C to 8°C storage, the updated expiry date must be written on
the outer carton and the vaccine should be used or discarded by the updated expiry date. The
original expiry date should be crossed out.
Check that the expiry date on the outer carton has been updated to reflect the refrigerated expiry
date and that the original expiry date has been crossed out.
under
When stored frozen at -90°C to -60°C, the vaccine can be thawed at either 2°C to 8°C or at
temperatures up to 30°C.
Prior to use, the unopened vials can be stored for up to 12 hours at temperatures between
8ºC to 30ºC.
the
Thawed vials can be handled in room light conditions.
Official
Once thawed COMIRNATY (orange cap, must dilute) should not be re-frozen.
Diluted medicinal product
Chemical and physical in-use stability has been demonstrated for 12 hours at 2ºC to 30°C, after
dilution with sodium chloride 9 mg/mL (0.9%) solution for injection. From a microbiological
Information
point of view, unless the method of dilution precludes the risk of microbial contamination, the
product should be used immediately. If not used immediately, in-use storage times and
conditions are the responsibility of the user.
6.4 Special precautions for storage
COMIRNATY (orange cap, must dilute) can be stored in a refrigerator at 2°C to 8°C for a
single period of up to 10 weeks, not exceeding the original expiry date (EXP). The expiry date
for storage at -90°C to -60°C is printed on the vial and outer carton after “EXP”.
Act
Check that the expiry date has been updated to reflect the refrigerated EXP date and that the
original expiry date has been crossed out.
1982
Store in the original package to protect from light. During storage, minimise exposure to room
light, and avoid exposure to direct sunlight and ultraviolet light.
For detailed instructions see Section 6.6 Special precautions for disposal and other handling.
Once thawed, the vaccine cannot be re-frozen.
Thawed vials can be handled in room light conditions.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 25 of 32
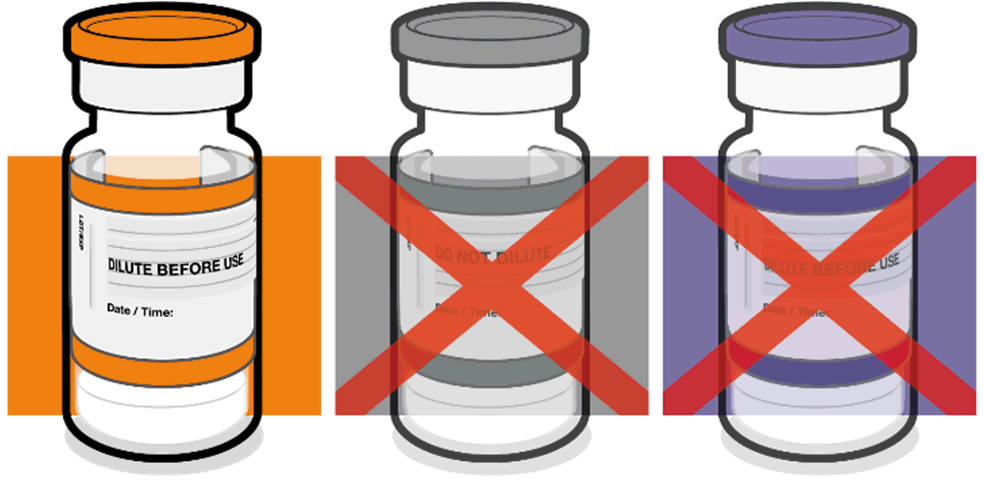


For storage conditions after thawing and dilution of the medicinal product, see Section 6.3
Shelf life.
For additional advice on storing COMIRNATY, contact Pfizer New Zealand on 0800 736 363.
6.5 Nature and contents of container
Released
COMIRNATY (orange cap, must dilute) 1.3 mL fill volume in 2 mL clear multidose vial
(Type I glass) with a stopper (synthetic bromobutyl rubber) and an orange flip-off plastic cap
with aluminium seal. Each vial contains 10 doses, see Section 6.6 Special precautions for
disposal and other handling.
Pack size: 10 vials, 195 vials
Not all pack sizes may be marketed.
under
6.6 Special precautions for disposal and other handling
COMIRNATY (orange cap, must dilute)
the
The vaccine should be prepared by a healthcare professional using aseptic technique to ensure
the sterility of the prepared diluted suspension.
Official
COMIRNATY (orange cap, must dilute)
Dose Verification
• Verify that the vial has an orange
plastic cap.
Information
Orange cap
• Only the orange cap vial can be used
for children age 5 to 11 years.
10 micrograms
Act 1982
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 26 of 32
 COMIRNATY (orange cap, must dilute)
Handling Prior To Use
COMIRNATY (orange cap, must dilute)
Handling Prior To Use
• If the multidose vial is stored frozen
it must be thawed prior to use.
Frozen vials should be transferred to
Released
an environment of 2°C to 8°C to
thaw; a 10 vial pack may take
4 hours to thaw. Ensure vials are
Store for up to
completely thawed prior to use.
10 weeks at
• Unopened vials can be stored for up
2 °C to 8 °C
to 10 weeks at 2°C to 8°C within the
24 month shelf life.
• Alternatively, individual frozen vials
under
may be thawed for 30 minutes at
temperatures up to 30°C for
immediate use.
the Official
Information
Act 1982
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 27 of 32
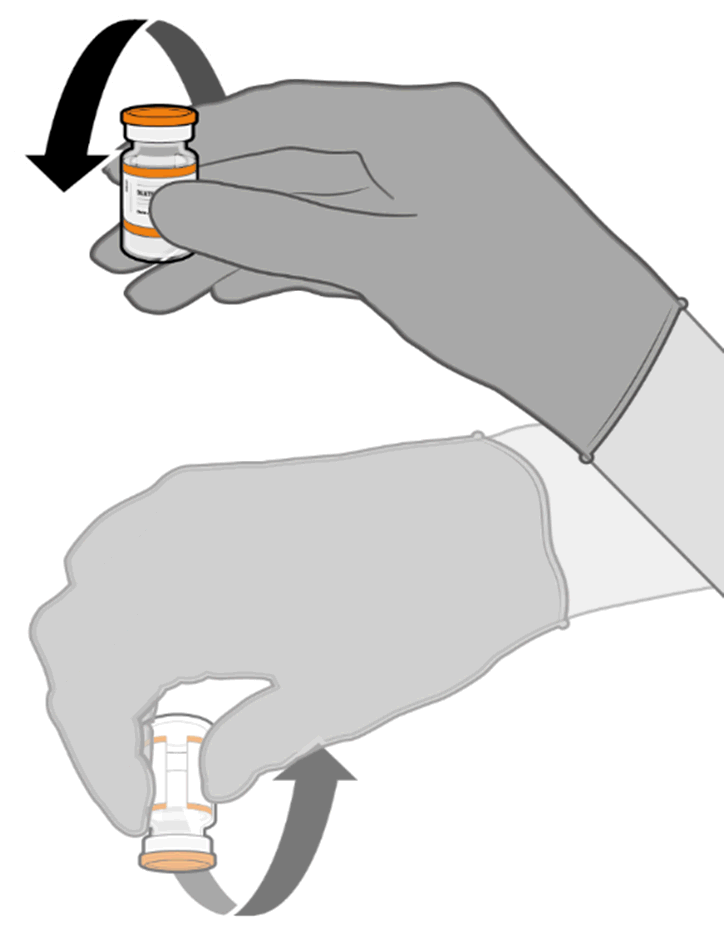
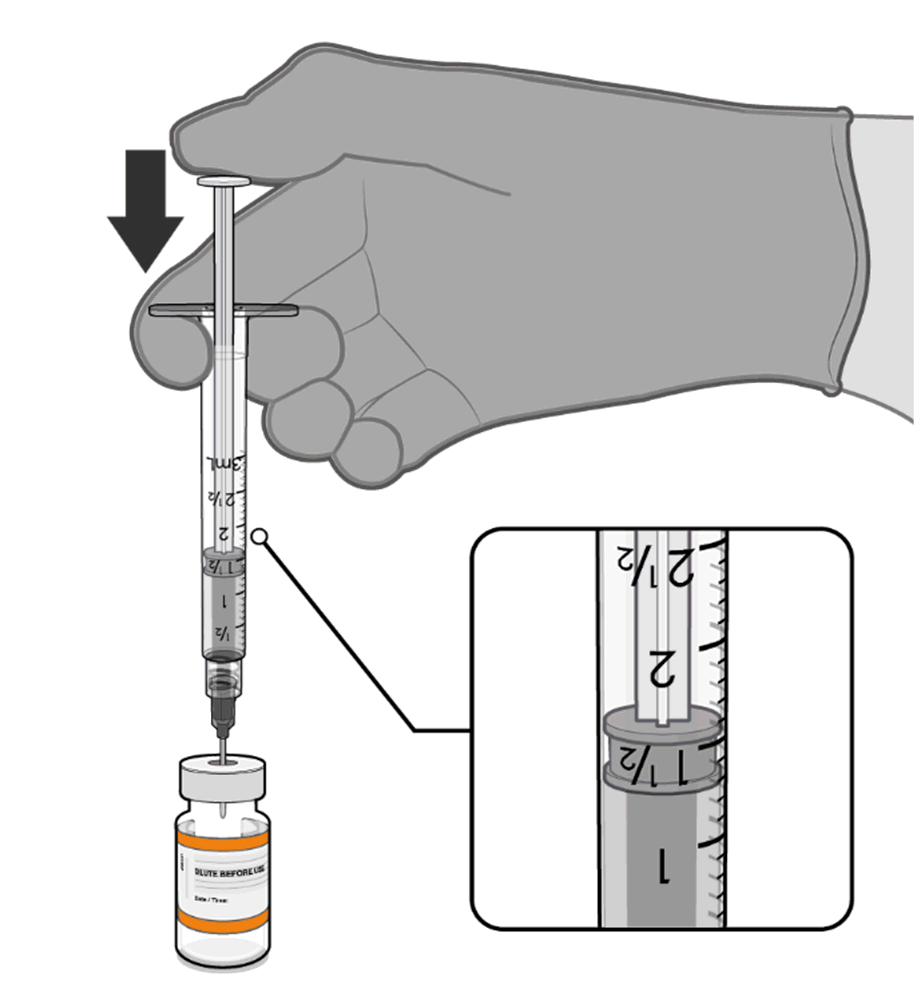 COMIRNATY (orange cap, must dilute)
Mixing Prior To Dilution
COMIRNATY (orange cap, must dilute)
Mixing Prior To Dilution
• Allow the thawed vial to come to
room temperature and gently invert it
10 times prior to dilution. Do not
Released
shake.
• Prior to dilution, the thawed
suspension may contain white to off-
white opaque amorphous particles.
under
the
Dilution
Official
• The thawed vaccine must be diluted
in its original vial with 1.3 mL
sodium chloride 9 mg/mL (0.9%)
solution for injection, using a
21 gauge or narrower ne
Information edle and
aseptic techniques.
Act 1982
1.3 mL of 0.9% sodium chloride
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 28 of 32
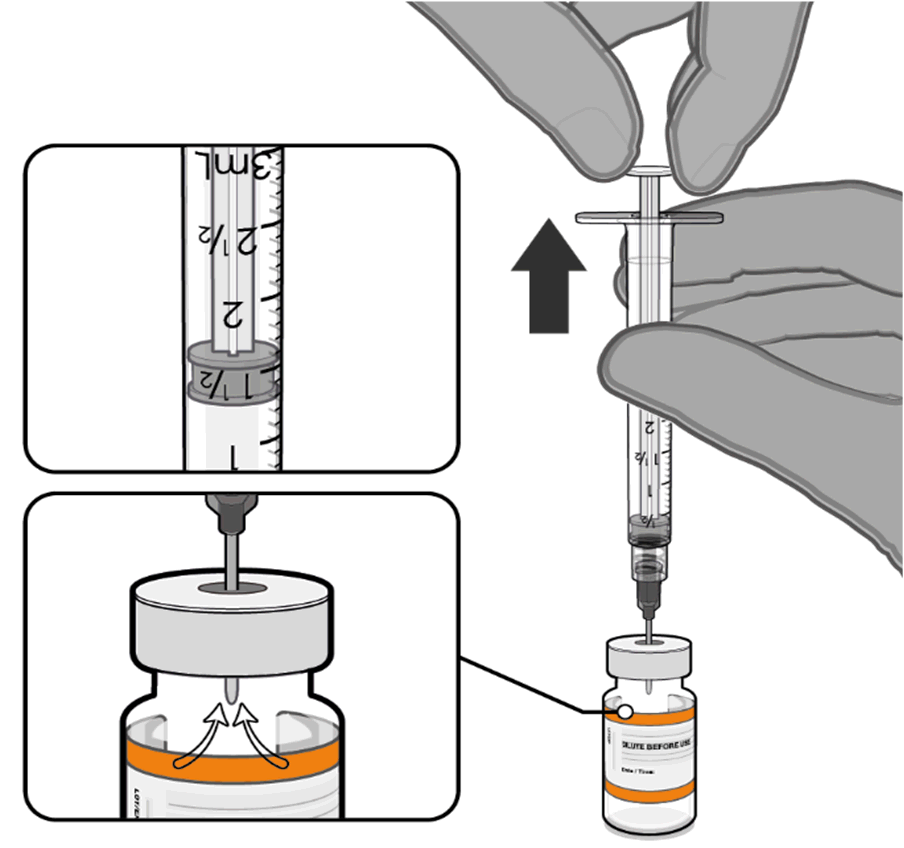
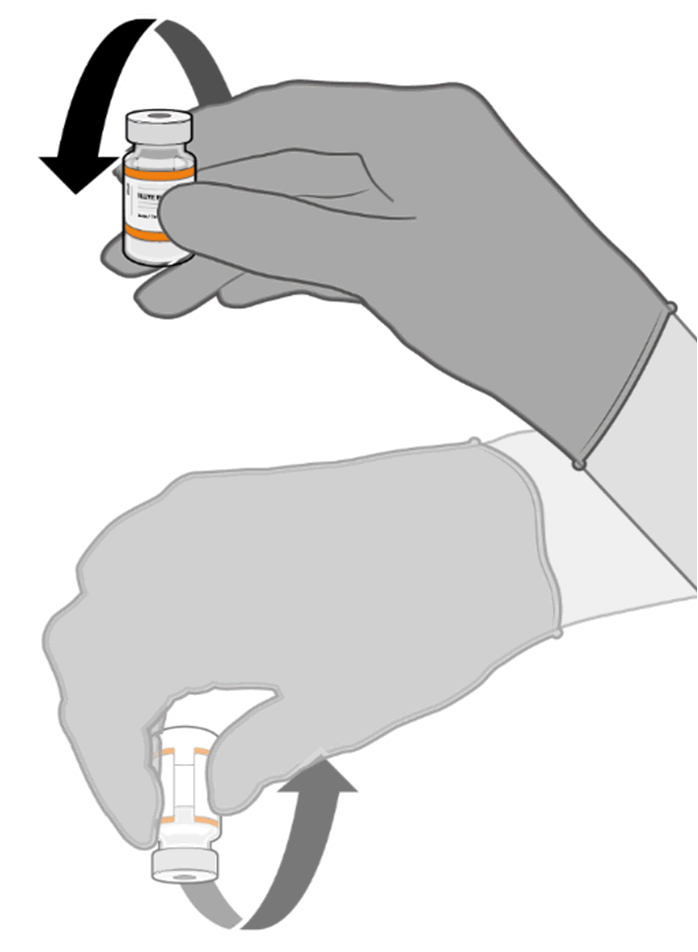 COMIRNATY (orange cap, must dilute)
Dilution (continued)
COMIRNATY (orange cap, must dilute)
Dilution (continued)
• Equalise vial pressure before
removing the needle from the vial
stopper by withdrawing 1.3 mL air
Released
into the empty diluent syringe.
under
the
Pull back plunger to 1.3 mL to
remove air from vial.
Official
• Gently invert the diluted suspension
10 times. Do not shake.
• The diluted vaccine should present as
a white to off-white suspension with
no particulates visible. Do not use
Information
the diluted vaccine if particulates or
discoloration are present.
Act 1982
Gently × 10
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 29 of 32
 COMIRNATY (orange cap, must dilute)
Dilution (continued)
COMIRNATY (orange cap, must dilute)
Dilution (continued)
• The diluted vials should be marked
with the appropriate date and time.
• After dilution, store at 2°C to 30°C
Released
and use within 12 hours.
• Do not freeze or shake the diluted
dispersion. If refrigerated, allow the
diluted suspension to come to room
temperature prior to use.
under
Record appropriate date and time.
Use within 12 hours after dilution.
the
Official
Information
Act 1982
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 30 of 32
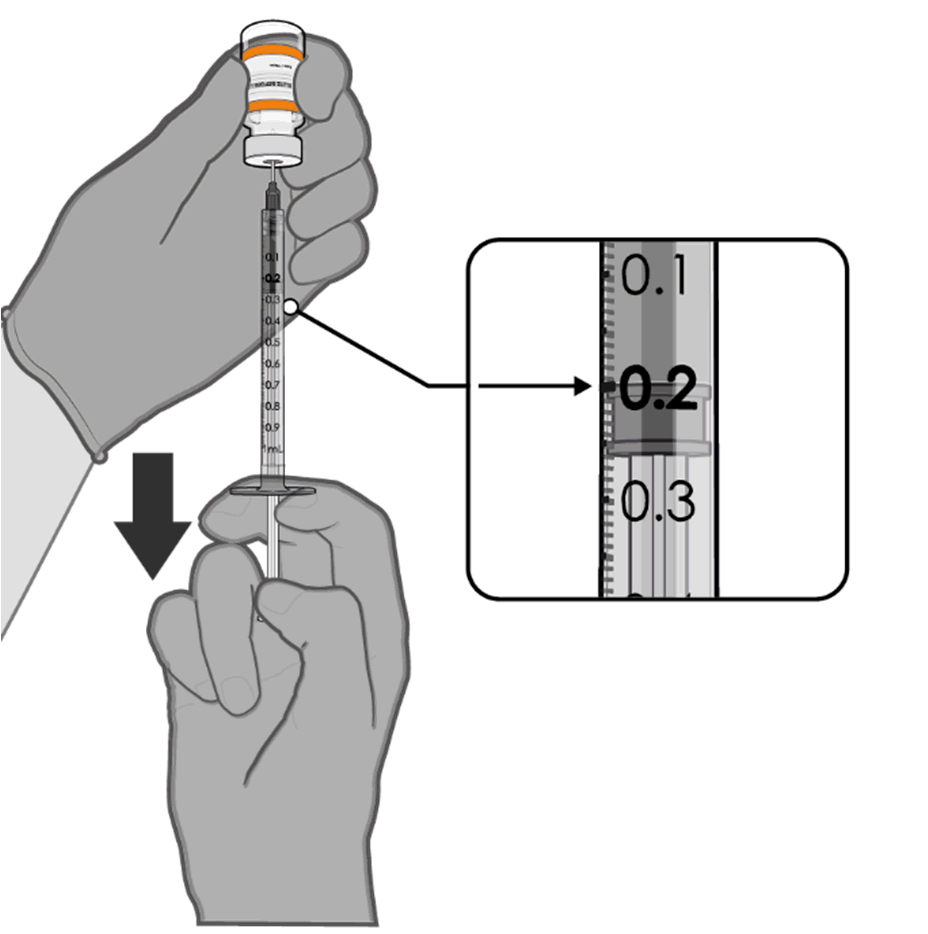 COMIRNATY (orange cap, must dilute)
Preparation of Individual 0.2 mL Doses of COMIRNATY (orange cap, must dilute)
COMIRNATY (orange cap, must dilute)
Preparation of Individual 0.2 mL Doses of COMIRNATY (orange cap, must dilute)
• Using aseptic technique, cleanse the
vial stopper with a single-use
antiseptic swab.
Released
• Withdraw 0.2 mL of COMIRNATY
(orange cap, must dilute).
Low dead-volume syringes and/or
needles should be used in order to
extract 10 doses from a single vial.
The low dead-volume syringe and
needle combination should have a
under
dead volume of no more than
35 microlitres.
If standard syringes and needles are
used, there may not be sufficient
the
volume to extract ten doses from a
single vial.
•
Official Each dose must contain 0.2 mL of
vaccine.
• Discard syringe and needle after
0.2 mL diluted vaccine
administration to a single patient.
• Use a new, sterile needle and syringe
to draw up each new dose.
Information
• If the amount of vaccine remaining
in the vial cannot provide a full dose
of 0.2 mL, discard the vial and any
excess volume.
• Discard any unused vaccine within
12 hours after dilution.
Any unused medicine or waste material should be disposed of in accordance with local
Act
requirements.
7. MEDICINE SCHEDULE
1982
Prescription Medicine.
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 31 of 32
8. SPONSOR
Pfizer New Zealand Limited
P O Box 3998
Auckland, New Zealand
Toll Free Number: 0800 736 363
Released
9. DATE OF FIRST APPROVAL
Date of publication in the New Zealand Gazette of consent to distribute this medicine:
16 December 2021
under
10. DATE OF REVISION OF THE TEXT
28 July 2023
COMIRNATY® is a registered trademark of BioNTech SE. Used under license.
the
Official
Summary of Updates
Section
Update
6.3
Update shelf life from 18 months to 24 months.
6.6
Update shelf life from 18 months to 24 months.
Information
Act 1982
Version: pfdcocii10723
Supersedes: pfdcocii21222
Page 32 of 32
Document Outline