Document 1
CLINICAL EVALUATION
Comirnaty
1982
(COVID-19 mRNA Vaccine)
ACT
Applicant: Pfizer/BioNTech
INFORMATION
OFFICIAL
Based on Final Analysis Interim Report - data cutoff: 14 November 2020
THE
TT Number
TT50-10853
Application ID
109400
Date received:
13 November 2020
UNDER
Date of this report:
January 2021
Evaluator
s 9(2)(g)(ii)
Reviewer
s 9(2)(g)(ii)
RELEASED
link to page 3 link to page 6 link to page 7 link to page 7 link to page 8 link to page 8 link to page 9 link to page 11 link to page 12 link to page 12 link to page 12 link to page 13
Document 1
TABLE OF CONTENTS
I.
INTRODUCTION ........................................................................................................................... 3
I.1
GCP aspects .................................................................................................................................. 6
II.
STUDY PROGRAMME ................................................................................................................. 6
II.1
Pharmacokinetics ......................................................................................................................... 6
II.1.1
First-in human study BNT162-01 ...................................................................................... 6
II.1.2
Introduction ............................................................................................................................ 9
II.2
First-in-human study BNT162-01............................................................................................ 10
III.
PHASE 1/2/3 STUDY C4591001 .............................................................................................. 10
III.1.1
Introduction .......................................................................................................................... 10
III.1.2
Phase 1 portion ................................................................................................................... 11
III.1.3
Candidate selection ........................................................................................................... 14
1982
III.2
Pivotal phase 2/3 Study C4591001 ......................................................................................... 16
IV.
CLINICAL EFFICACY PHASE 2/3 STUDY C4591001 .......................................................... 19
IV.1
Dose-response studies and main clinical studies .............................................................
ACT 19
IV.1.1
Dose selection ..................................................................................................................... 19
IV.1.2
Main study: Phase 2/3 Study C4591001 14 Nov 2020 cutoff ................................... 20
IV.2
Final analysis phase 2/3 ........................................................................................................... 30
IV.3
Subgroup analysis ..................................................................................................................... 37
IV.4
Signs and symptoms of COVID-19 ........................................................................................ 44
IV.5
Evaluator’s overall conclusions on clinical efficacy ........................................................ 47
V.
CLINICAL SAFETY PHASE 2/3 STUDY C4591001 .............................................................. 48
V.1
Introduction .................................................................................................................................. 48
INFORMATION
V.2
Patient exposure ......................................................................................................................... 49
V.2.1
Population for safety analysis ........................................................................................ 51
V.3
Phase 2/3 reactogenicity subset ............................................................................................ 52
V.3.1
Reactogenicity ..................................................................................................................... 52
V.4
Adverse events ........................................................................................................................... 59
V.5
Serious adverse events and deaths ...................................................................................... 64
OFFICIAL
V.6
Proposals for post authorisation follow up (post marketing surveillance) ................ 67
V.6.1
EUA Safety Surveillance Study Plan ............................................................................. 67
V.6.2
Pharmacovigilance Plan ................................................................................................... 67
THE
V.7
Post marketing experience/MHRA notification 08 December 2020 ............................... 68
V.8
Post marketing experience/Norway deaths ......................................................................... 70
V.9
Evaluator’s overall conclusions on clinical safety ............................................................ 70
VI.
benefit risk assessment .............................................................................................................. 71
VII.
PRODUCT INFORMATION ....................................................................................................... 73
UNDER
VIII.
recommendation .......................................................................................................................... 74
IX.
SELECTED INITIAL MAAC COMMENTS ............................................................................... 74
I.
QUESTIONS and applicant’s response ................................................................................... 74
II.
SUMMARY ................................................................................................................................... 78
RELEASED
Limited glossary
ACV
TGA’s Advisory Committee on Vaccines
CEF
MHC-class I restricted peptides originating from CMV, EBV, and
flu (influenza) virus
CEFT
MHC-class II restricted peptides originating from CMV, EBV, Flu
(influenza) virus and tetanus toxin
Document 1
CoV
coronavirus
COVID-19
coronavirus disease 2019
CSR
clinical study report
EUA
(FDA’s) Emergency Use Authorization
FACS
fluorescence-activated cell sorting
HCS
Convalescent human serum
IA
interim analysis
IRC
internal review committee
1982
IRR
illness rate ratio
IWR
interactive Web-based response
ACT
LNP
lipid nanoparticle
MERS
Middle East respiratory syndrome
modRNA
nucleoside-modified messenger ribonucleic acid
NAAT
nucleic acid amplification test
N-binding
SARS-CoV-2 nucleoprotein binding
NT50
neutralizing titer 50
INFORMATION
NT90
neutralizing titer 90
NVA
nonvaccine antigen
P2 S
SARS-CoV-2 full-length, P2 mutant, prefusion spike glycoprotein
P/B
Dose 1/Dose 2: a dosing regimen, comprising a priming
immunization (dose 1) and a dose 2 immunization (dose 2)
OFFICIAL
PBMCs
peripheral blood mononuclear cells
RBD
receptor-binding domain
THE
SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
SIRVA
shoulder injury related to vaccine administration
SRC
Safety Review Committee
UNDER
Tdap
diphtheria vaccine toxoid; pertussis vaccine acellular 3 component;
tetanus vaccine toxoid
RELEASED
SUMMARY REPORT
I.
INTRODUCTION
Pfizer have submitted a new medicine application, received 13 November 2021 (ID 109400), for
a nucleoside modified messenger RNA (modRNA) vaccine: the Pfizer-BioNTech COVID-19
Vaccine (BNT162b2 30 µg): Comirnaty (COVID-19 mRNA Vaccine).
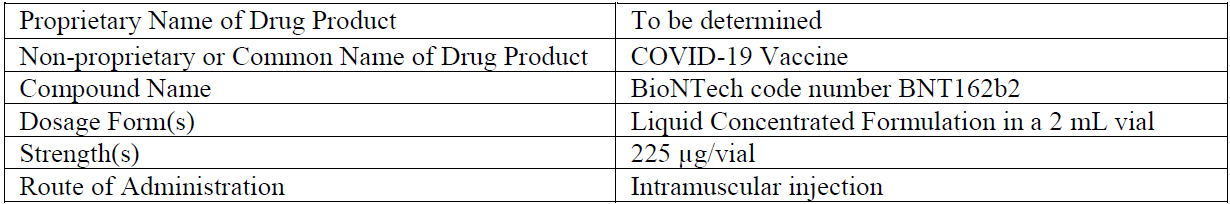
Document 1
Comirnaty (30 μg), is administered intramuscularly (IM) as a series of two 30-μg doses of the
diluted vaccine solution (0.3 mL each) according to the following schedule: a single dose
followed by a second dose 21 days later.
The proposed indication is as follows.
COMIRNATY is indicated for active immunisation to prevent coronavirus disease 2019
(COVID-19) caused by SARS-CoV-2, in individuals 16 years of age and older.
The use of this vaccine should be in accordance with official recommendations.
Evaluator’s comment
The TGA’s Delegate’s Overview shows that the ‘Indication revised by Sponsor
following TGA request’ is as follows:
1982
COMIRNATY (BNT162b2[mRNA]) COVID-19 Vaccine has
provisional
approval for the indication below:
Active immunisation to prevent coronavirus disease 2019 (COVID-19)
ACT
caused by SARS-CoV-2, in individuals 16 years of age and older.
The use of this vaccine should be in accordance with official
recommendations.
The decision has been made on the basis of short term efficacy and safety
data. Continued approval depends on the evidence of longer term efficacy
and safety from ongoing clinical trials and post-market assessment.
This vaccine encodes P2 S (V9), expresses a prefusion stabilized full-length variant of the
INFORMATION
SARS-CoV-2 S-glycoprotein.
The RNA-based vaccine is formulated in lipid nanoparticles (LNPs) and includes two novel lipid
excipients.
OFFICIAL
THE
Formulation
The vaccine candidate will be released as a concentrated multi-dose liquid formulation stored
frozen at -90 to -60 °C in a 2 mL Type 1 glass vial to be thawed and subsequently diluted with
UNDER
sterile 0.9% sodium chloride Solution for Injection, USP (saline diluent), and stored at 2-8 °C
until administration.
Dose and administration
The vaccine will be administered intramuscularly (IM) in the upper arm (deltoid muscle) as a
series of two 30 μg doses of the diluted vaccine solution (0.3 mL each) according to the
RELEASED
following schedule: a single 0.3 mL dose followed by a second 0.3 mL dose 21 days later
(prime/boost regimen).
The draft datasheet includes, among other details, that the vaccine comes as concentrated
suspension for injection for 5 doses in a 2 mL clear vial.
Store in a freezer at -90 °C to -60 °C. After thawing, the vaccine should be diluted and used
immediately. After dilution, store the vaccine at 2 °C to 30 °C and use within 6 hours.
 1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
 1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
Document 1
For participants without evidence of SARS-CoV-2 infection before and during vaccination
regimen, from 7 days after the second dose, there were the following cases of symptomatic
laboratory-confirmed COVID-19 of any severity;
▪
8
(out of 18,198; 0.04%) in the BNT162b2 group and
▪
162
(out of 18,325; 0.9%) in the placebo group.
In the placebo group, 162 instances of symptomatic COVID infection in about 2,222 person-
years. Given the approximately 18,000 subjects who received placebo, surveillance time for the
majority of subjects is likely to be between one to two months.
For the other co-primary efficacy endpoint, VE against confirmed COVID-19 in participants with
or without evidence of SARS-CoV-2 infection was 94.6% (with 9 and 169 cases in the
1982
BNT162b2 and placebo groups respectively). In the elderly, participants ≥65 years of age with
or without prior evidence of SARS-CoV-2 infection, VE was 94.7% (corresponding to 1 case in
the BNT162b2 and 19 in the placebo groups).
ACT
‘Severe’ confirmed COVID-19 meant that subject had in addition to the confirmed Covid-19 (for
example) at least; severe systemic illness (eg RR ≥30 breaths per minute); or needing high-flow
oxygen, or admission to an ICU; or death. Severe disease was noted in one case of the
vaccinated group and 3 cases in the placebo group. Although at this stage of the study’s follow-
up, only about 1% of placebo subjects have developed symptomatic COVID-19, severe disease
was not common (about 2% of those with symptomatic disease had severe disease; 3 out of
162).
Some subjects who at baseline had evidence of prior COVID-19 infection, subsequently
INFORMATION
developed symptomatic COVID-19 at least 7 days after Dose 2.
As noted in the VRBPAC
Briefing Document, only 3% of participants had evidence of prior infection at study enrolment.
These data do suggest that previously infected individuals can be at risk of COVID-19 (i.e.,
reinfection) and could benefit from vaccination.
V.
s 9(2)(b)(ii)
OFFICIAL
s 9(2)(b)(ii)
THE
UNDER
RELEASED
Pages 49- 69 withheld under section 9(2)(b)(ii) of the Act.
Document 1
s 9(2)(b)(ii)
V.8
Post marketing experience/Norway deaths
There are reports of deaths of 23 frail elderly patients shortly after receiving the Pfizer BioNTec
vaccine. The Norwegian Medicines Agency (NOMA) has commented that there is no certain
connection between these deaths and the vaccine.
The agency has investigated 13 of the deaths so far and concluded that common adverse
1982
reactions of mRNA vaccines, such as fever, nausea, and diarrhoea, may have contributed to
fatal outcomes in some of the frail patients. “There is a possibility that these common adverse
reactions, that are not dangerous in fitter, younger patients and are not unusual with vaccines,
ACT
may aggravate underlying disease in the elderly”.
Norwegian Authorities have prioritized the immunization of residents in Nursing Homes, most of
whom are very elderly with underlying medical conditions and some which are terminally ill.
NOMA confirms the number of incidents so far is not alarming, and in line with expectations.
All reported deaths will be thoroughly evaluated by NOMA to determine if these incidents are
related to the vaccine. The Norwegian government will also consider adjusting their vaccination
instructions to take the patients’ health into more consideration.
https://www.bmj.com/content/372/bmj.n149
INFORMATION
News. Covid-19: Norway investigates 23 deaths in frail elderly patients after vaccination
BMJ 2021; 372 doi: https://doi.org/10.1136/bmj.n149 (Published 15 January 2021)Cite this
as: BMJ 2021;372:n149
OFFICIAL
V.9
Evaluator’s overall conclusions on clinical safety
In the Phase 2/3 Study C4591001 subjects were randomised to receive the modRNA COVID-19
THE
Vaccine (BNT162b2 30 µg) three weeks apart. As at data cutoff 14 November 2020, safety
information is available for the ‘all subjects’ safety population N~38,000 with medium follow-up of
two months. The safety population with at least 2 months of follow-up after dose 2 had n =
19,067.
The vaccine is reactogenic, which is e
UNDER vident especially through information from the Phase 2/3
reactogenicity subset (using e-diary reporting). As per the CHMP assessment report summary:
“Regarding reactogenicity, the most frequent adverse reactions in participants 16 years of age
and older were injection site pain (> 80%), fatigue (> 60%), headache (> 50%), myalgia and
chills (> 30%), arthralgia (> 20%), pyrexia and injection site swelling (> 10%). All reactions were
usually mild or moderate in intensity and resolved within a few days after vaccination. A slightly
lower frequency of reactogenicity events was associated with greater age. The frequency of
headache, fatigue and
RELEASED fever was higher after Dose 2 in both age groups.”
For participants who were not in the reactogenicity subset, local reactions and systemic events
consistent with reactogenicity were detected and reported as AEs. AEs judged to be related to
vaccination were noted in 21% of BNT162b recipients vs 5% in the placebo group. This
includes injection site pain (7%), pyrexia (4%), as well as chills, fatigue, headache, and myalgia
at lesser levels.
Document 1
AEs were usually reported at a higher level in the younger (18-55 Years of Age) age group than
in the older (65-85 Years of Age) age group. For example, 31% of this younger age group had
moderate pain after the first dose, and 27% after the second dose. Systemic symptoms were
common especially following the second dose (eg fatigue, headache, muscle pain and joint
pain). Fever of ≥38.0°C after the second dose was noted in the younger group by 15.8%, and
of ≥38.0°C to 38.4°C by 9.2%.
However, severe AEs were reported in 1.2% of the vaccine group compared to 0.6% of the
(saline) placebo group, and AEs leading to withdrawal 0.2% vs 0.1%.
Uncommonly reported was lymphadenopathy in the arm and neck region (0.5% in the younger
group). This was reported within 2 to 4 days after vaccination, and sometimes was slow to
resolve. In addition, one subject reported angioedema 13 days after Dose 1 affecting both eyes,
1982
and another subject report hypersensitivity (allergy attack).
There was an imbalance of cases of Bell’s palsy (4 in the vaccine group and none in the placebo
ACT
group).
VI.
BENEFIT RISK ASSESSMENT
Covid-19 pandemic
In early 2021 it is becoming clear that the novel coronavirus SARS-CoV-2 constitutes an
important health hazard, especially for the elderly as well as people with comorbidities. Even
relatively low mortality rates associated with the resulting disease, COVID-19, may have a
substantial impact as the whole population is assumed to be susceptibility. There have now
INFORMATION
been months of waves of increased transmission and disease. In addition to the immediate
sickness, a relevant proportion of patients suffer longer term adverse consequences; including
eg respiratory and cardiovascular system impairment, as well as long-Covid syndrome.
Medical need
Public health measures have been shown to be potentially very effective, although such
OFFICIAL
measures can be socially disruptive and can have large economic consequences.
Treatment of acute Covid-19 disease has improved, and several medicines are recognised to
THE
have a role in treatment.
Populations for benefit risk assessment
The probability of exposure to the virus, as well as the mortality and morbidity burden associated
with the disease is relevant to the benefit risk. Arguably, the following populations could be
UNDER
considered for separate benefit risk assessments.
▪
The benefit risk balance of a COVID-19 Vaccine as a travel vaccine for the elderly would
likely be positive for many vaccines with reasonable efficacy and safety, as globally the
virus is now endemic and chance of exposure is high.
▪
For New Zealand residents, staff at quarantine facilities, as well as Air New Zealand staff
working on international routes and healthcare professionals, are at increased risk of
RELEASED
contact with the virus.
▪
As long as public health measures continue to be effective, vaccination of New
Zealanders generally could become relevant when vaccine supplies allow for the entire
New Zealand resident high-risk population to be covered.
Vaccine characteristics
Document 1
The vaccine’s preservative free multiple dose presentation, with need for administration close to
low-temperature storage, will likely result in use in group-settings rather than episodic individual
use setting (such as relevant for a travel-vaccine). Given the LNP-mRNA vaccine innovative
technology, particular care in the evaluation of safety (including longer-term safety) is important.
Benefit
The randomised Phase 2/3 Study C4591001 shows that for participants without evidence of
SARS-CoV-2 infection before and during vaccination regimen, from 7 days after the second
dose, there were the following cases of symptomatic laboratory-confirmed COVID-19 of any
severity:
▪
8
(out of 18,198; 0.04%) in the BNT162b2 group and
1982
▪
162
(out of 18,325; 0.9%) in the placebo group.
With currently available data from the pivotal study, follow-up data is only available for about one
to two months. Therefore there is uncertainty about how long protection will last. At the earliest,
ACT
updated efficacy estimates regarding longer duration of vaccine protection are expected to
become available from April 2021.
In addition, the number of cases of symptomatic COVID-19 in subgroups of the study population
is often low. There is thus uncertainty regarding efficacy in people of Polynesian and Asian
ethnicity.
Severe disease was noted in one case of the vaccinated group and 3 cases in the placebo
group; an estimated efficacy against severe COVID-19 occurring at least 7 days after dose 2
was 66.4% (95% CI: -124.8%; 96.3%).
INFORMATION
Risk
The vaccine is reactogenic, which is shown especially through information from the Phase 2/3
reactogenicity subset (using e-diary reporting), for example: injection site pain (> 80%), myalgia
and chills (> 30%), and pyrexia and injection site swelling (> 10%) are very common. There are
lesser rates when reporting of AEs is considered, for example general disorders and
OFFICIAL
administration site conditions (12% BNT162b2 vs 3% placebo). In general, AEs judged to be
related to vaccination were noted in 21% of BNT162b recipients vs 5% in the placebo group -
THE
AEs includes injection site pain (7%), pyrexia (4%), as well as chills, fatigue, headache, and
myalgia at lesser levels. Severe AEs were reported in 1.2% of the vaccine group compared to
0.6% of the (saline) placebo group, and AEs leading to withdrawal 0.2% vs 0.1%. Longer-term
safety data is lacking.
AEs were usually reported at a higher level in the younger (18-55 Years of Age) age group than
UNDER
in the older (65-85 Years of Age) age group. For example, 31% of this younger age group had
moderate pain after the first dose, and 27% after the second dose. Systemic symptoms were
common especially following the second dose. Fever of ≥38.0°C after the second dose was
noted by about 16% in the younger group.
Uncertainties
Pivotal trial design and sample size means that study results are not expected to address all of
RELEASED
the following uncertainties.
▪
It is not clear that the method of administration of the Comirnaty vaccine, as described in
the datasheet’s ‘Special precautions for disposal and other handling’ section, is similar to
the method of administration in the pivotal study.
▪
The duration of vaccine protection has not been established beyond two months.
▪
At this stage, there is limited evidence of protection against severe disease.
Document 1
▪
There is no long-term safety follow-up information.
▪
Vaccine prevention of asymptomatic infection and disease transmission has not been
established.
At this stage there is no information regarding vaccine effectiveness regarding:
▪
new variant virus lineages that may become important epidemiologically (including the
possibility of change because of vaccine-selection pressures)
▪
immunocompromised people, and for pregnant women
▪
Pacific and Asian populations
▪
subjects with evidence of prior COVID-19 infection at baseline.
1982
Summary
The benefit risk balance of Comirnaty (COVID-19 mRNA Vaccine) for active immunisation to
ACT
prevent coronavirus disease 2019 (COVID-19) caused by SARS-CoV-2, in individuals 16 years
of age and older, is not clear. At this stage, there is evidence only for short-term protection, and
longer-term safety data are lacking. However, experience with the vaccine is accumulating
rapidly.
Notwithstanding uncertainties, in the light of high clinical need and the expectation of further
data (including regarding duration of protection) around April 2021, a provisional consent under
section 23 of the Medicines Act 1981 may be appropriate.
INFORMATION
VII.
PRODUCT INFORMATION
If considered for provisional consent, the datasheet will have to explain that approval was based
on short-term vaccine-protection information, and that further information is expected.
Among others, issues covered in the datasheet include the following.
The study programme did not cover pregnant women and
OFFICIAL children.
Pregnancy
THE
There is limited experience with use of COMIRNATY in pregnant women. Animal studies
do not indicate direct or indirect harmful effects with respect to pregnancy, embryo/fetal
development, parturition or post-natal development (see Section 5.3 Preclinical safety
data).
Administration of COMIRNATY in pregnancy should only be considered when the
UNDER
potential benefits outweigh any potential risks for the mother and fetus.
Evaluator’s comment
s 6(b)(ii)
It is often advantageous for the New Zealand
datasheet to be similar to the information for prescribers in Australia, and
RELEASED
updating the pregnancy information in the proposed New Zealand datasheet to be
similar to that for Australia should be considered.
IM administration
Thrombocytopenia and coagulation disorders
As with other intramuscular injections, COMIRNATY should be given with caution in
individuals receiving anticoagulant therapy or those with thrombocytopenia or any

Document 1
coagulation disorder (such as haemophilia) because bleeding or bruising may occur
following an intramuscular administration in these individuals.
Hypersensitivity
1982
ACT
Evaluator’s comment
For justification of the above general recommendation, see the section in the
Safety part of this report regarding the MHRA notification of two allergic reaction
events in UK December 2020.
VIII.
RECOMMENDATION
INFORMATION
The application is to be referred to the MAAC for final recommendation on provisional consent,
as well as the proposed conditions.
IX.
SELECTED INITIAL ADVISORY GROUP COMMENTS
Responses to an early request (with very limited information) for advice from the Medsafe
OFFICIAL
COVID-19 Vaccine Advisory Committee have included the following.
Covid-19 vaccines can be expected not to provide long term protection – the need for booster
THE
doses can be expected. (For viral vectored vaccines, heterologous boosting may be needed).
Significant delayed adverse consequences of vaccination, generally, are very uncommon. For
example, a recent article highlighted vaccines that had been withdrawn for safety concerns. All
of the events, resulting in withdrawal, occurred within 2 months of vaccine receipt (Reid S
UNDER
Vaccine Safety NZMJ 21 February 2020 Vol 133 No 1510. www.nzma.org.nz/journal-
articles/vaccine-safety). Possible delayed AEs could include:
▪
VAERD in specific age groups (eg geriatric, pediatric) or in individuals with uncommon
comorbidities (eg autoimmunity / immune deficiency)
▪
Guillain Barre Syndrome
▪
narcolepsy.
RELEASED
s 9(2)(b)(ii)
Pages 75- 77 withheld under section 9(2)(b)(ii) of the Act.
Document 1
s 9(2)(b)(ii)
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
II.
SUMMARY
Comirnaty COVID-19 mRNA vaccine
Document 1
This application concerns the Pfizer-BioNTech nucleoside modified messenger RNA vaccine,
Comirnaty COVID-19 mRNA vaccine that is administered as two intramuscularly injected doses,
21 days apart. This innovative lipid nanoparticle RNA-based vaccine includes two novel lipid
excipients. Administration is made more challenging by its availability as a concentrated multi-
dose liquid formulation stored frozen at -90 to -60 °C.
Analysis of condition and current treatment options
Although treatments have improved, COVID-19 causes substantial morbidity and mortality in a
susceptible population. Vaccination can mitigate the impact of COVID-19.
Benefits
The pivotal study shows two doses the Comirnaty COVID-19 mRNA vaccine (BNT162b2 30 µg)
1982
three weeks apart to provide a high level (95%) of protection against symptomatic COVID-19 (as
at data cutoff 14 November 2020, n≈ 37,000 with follow-up usually about one to two months).
Uncertainty
ACT
There is uncertainty regarding the duration of protection. In addition, it is not known whether
vaccinated people can become infected asymptomatically and whether they can transmit the
virus. At this stage, it is not known whether the vaccine protects against severe disease, and
whether it would provide protection for subgroups (such as, for example, the elderly).
The Sponsor, in response to the Request for Information, noted that efficacy data is expected in
2021.
▪
Q1 2021; initial results regarding the possibility of asymptomatic infection in the
INFORMATION
vaccinated group.
▪
First half of 2021; Information regarding vaccine failure in patients given 2 doses of
vaccine.
▪
Q3 2021; immunogenicity data for a subset of participants.
▪
Q3-4 2021; information regarding duration of vaccine protection.
OFFICIAL
If considered for provisional consent, the datasheet could (as is not uncommon) harmonise with
the TGA approved product information and have an indication that explains that approval was
THE
based on short-term vaccine-protection information, and that further information is expected.
Risks
In the study programme, safety of the Comirnaty COVID-19 mRNA vaccine was similar to that of
IM administered vaccines.
UNDER
Uncertainty
While there is uncertainty regarding the unlikely possibility of rare or delayed AEs, there is
increasing assurance of the safety of the Comirnaty COVID-19 mRNA vaccine with rapidly
increasing international experience. Monthly periodic safety update reports will be expected for
the first 6 months post approval.
RELEASED
II.1
Advice sought
In its consideration of this application, the Medicines Assessment Advisory Committee (MAAC)
is asked to advise whether:
−
The proposed conditions for the provisional consent are appropriate.
Document 1
−
The benefit risk balance of the Comirnaty COVID-19 mRNA vaccine for active
immunisation to prevent coronavirus disease 2019 (COVID-19) caused by SARS-CoV-2,
in individuals 16 years of age and older is positive.
−
Whether to accept the proposed indications, or to request that the Sponsor update the
New Zealand datasheet so that it is harmonised with the TGA approved product
information, and includes the following indication:
Comirnaty (BNT162b2 (mRNA)) COVID-19 vaccine has
provisional consent for
the indication below:
Active immunisation to prevent coronavirus disease 2019 (COVID-19)
caused by SARS-CoV-2, in individuals 16 years of age and older.
1982
The use of this vaccine should be in accordance with official
recommendations.
The decision has been made on the basis of short-term efficacy and
ACT
safety data. Continued approval depends on the evidence of longer-term
efficacy and safety from ongoing clinical trials and post-market
assessment.
II.2
Proposed conditions of provisional consent
▪
The sponsor must provide regular updates on the duration of efficacy and the
requirement for booster doses. This should include the six months analysis data from
INFORMATION
Study C4591001. Interim report due: April 2021.
▪
The sponsor must provide updates on efficacy including regarding asymptomatic
infection in the vaccinated group, vaccine failure, immunogenicity, efficacy in population
subgroups and results from post-marketing studies, when these become available.
▪
The sponsor must submit the final Clinical Study Reports for Study C4591001 and Study
OFFICIAL
BNT162-01. Due date: December 2023.
▪
(Monthly safety updates for the first six months following approval are covered through
the RMP.)
THE
UNDER
RELEASED
 1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
 1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25
Document 2
Evidence summary: transmissibility of SARS-CoV-2,
particularly new Variants of Concern (VOC)
Introduction
All viruses constantly change through mutation, and therefore the emergence of new variants is expected
(1).
Some mutations do not confer a direct benefit to the virus, some may be detrimental, and some may confer
an advantage
(2). New variants may emerge and disappear, or they may persist
(3). Most mutatio
1982ns won’t
significantly impact viral spread, but some mutations or combinations of mutations may give viruses a
selective advantage (e.g. increased transmissibility due to an increase in receptor binding, or evasion of the
ACT
host immune response by altering viral surface structures)
(1). Many thousands of variants of SARS-CoV-2 are
circulating, and most will likely have no effect on viral transmission or disease characteristics
(1). Variants with
potential to increase the risk to human health are considered variants of concern (VOC)
(1).
ation Act 1982
Multiple variants of SARS-CoV-2 have been documented globally throughout the course of the pandemic
(3),
but some recent variants are causing particular alarm because of reported increases in transmissibility. Three
variants of note are discussed in this paper, which appear to have emerged in the UK (lineage B.1.1.7), South
Africa (501.V2) and Brazil (P.1). Lineage B.1.1.7 (UK variant) is discussed in most
INFORMATION detail as it has more
information available. Scientists are working to figure out whether new variants such as these may transmit
more easily, cause more severe disease, or affect the efficacy of therapeutics or vaccines
(3). Epidemiologically,
it is quite difficult to distinguish contributors to more efficient spread (e.g. human behavioural factors, vs
virologic factors)
(4).
OFFICIAL
This evidence summary provides background information about the transmissibility of SARS-CoV-2 in
general, and the UK variant in particular. It also considers potential implications.
THE
A note on naming conventions: When a number of mutations occur together and become commonly
detected, they are designated as a lineage or variant. A viral strain is typically a substantial change in
under the O
the virus – the consensus is that none of the SARS-CoV-2 lineages are yet at a point where they are
UNDER
designated as a formal strain.
Released
UK variant
RELEASED
Origin, discovery and spread
• In December 2020, a new variant of SARS-CoV-2 (called VOC-202012/01) was reported in the United
Kingdom
(5). It was designated a Variant Under Investigation on detection and re-designated as a
Variant of Concern (VOC) on 18 December 2020
(6). It is also called lineage B.1.1.7. This variant was
identified by the COVID-19 Genomics UK (COG-UK) consortium, which undertakes random genetic
sequencing of positive samples from around the UK to help track outbreaks and identify variants
(7).
The authors of the COG-UK report hypothesise that the accelerated mutation accumulation in this
lineage may have resulted, at least partly, from virus evolution within a chronically infected individual
link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25
Document 2
(8). However, they are careful to note we cannot yet know precisely what gave rise to this lineage.
Experts indicate it is likely to have evolved in the UK
(7).
• The first COVID-19 case with the VOC 202012/01 variant in England was detected on 20 September
2020. Throughout December 2020, a cluster of the new variant grew rapidly, and spread to other UK
locations
(8). The UK experienced a rapid increase in COVID-19 case rates (with the seven-day case
rate increasing from 162 cases/ 100,000 population in week 49/2020, to 344 during week 51/2020
(1).
This increased case rate was especially significant in London, the South East and the East of England,
and genomic analysis identified that a large proportion of sequenced cases in these areas were a new
variant, VOC 202012/01
(1). The rapid increase in COVID-19 cases overall was noted to be temporally
associated with the emergence of a new variant in the abovementioned areas in November 2
1982 020
(1).
The B.1.1.7 lineage rapidly emerged to become the dominant SARS-CoV-2 variant circulating in
England
(6).
ACT
• In response to the increase in VOC 202012/01, in late December the UK announced stricter control
measures to be applied, especially in affected areas in England
(1)
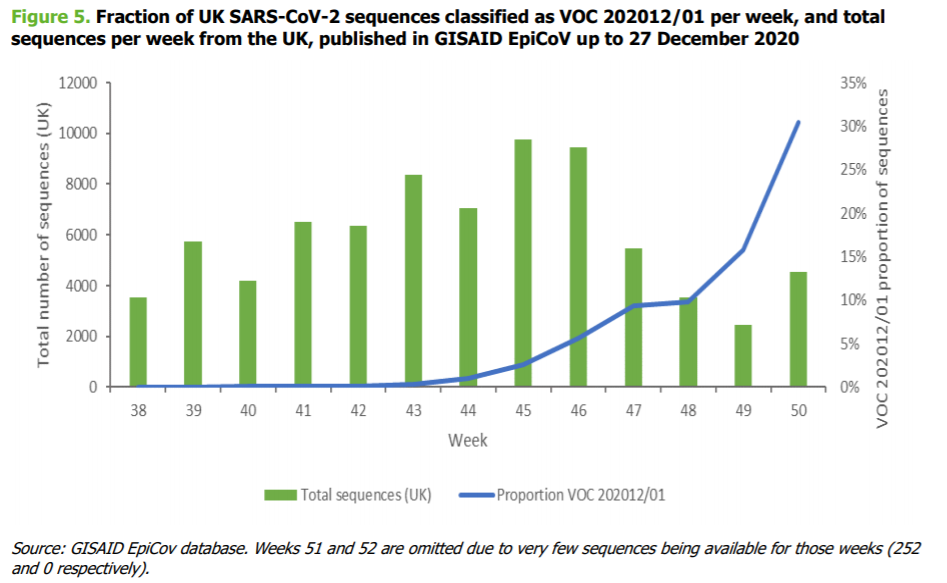
• As of 13 December 2020, VOC 202012/02 had been identified in 1108 individuals in the UK, in nearly
ation Act 1982
60 different local authorities
(7). The figure below from the ECDC risk assessment displays how the
overall proportion of VOC202012/01 among all uploaded viral sequences from the UK to the GISAID
database increased substantially towards the end of 2020. However, it should be noted that this data
is derived from community-based sampling, and is not geographicall
INFORMATION y representative, or
representative of hospitalised cases
(1).
OFFICIAL
THE
under the O
UNDER
Released
RELEASED
• As of 4 January 2021, a total of 6,008 cases with this variant had been identified in England, via routine
genomic surveillance, across the majority of local authorities
(7). Most cases were identified in London
and the East and South East of England but the variant has also been reported elsewhere, including
Wales and Scotland
(7).
• As of 7th Jan 2021, 45 countries had reported the presence of the B.1.1.7 variant
(5), including in
managed isolation in New Zealand. As of 18 January, 16 cases of the B.1.1.7 variant were reported in
New Zealand.
link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25
Document 2
• The B.1.1.7 variant is quickly becoming the dominant lineage across the UK, though it is unclear how
much this is due to viral genetics, versus seasonal changes and social factors. As of 4 January 2021,
the new variant comprises approximately 71.5% of new cases in the UK. However, the new variant may
not be simply replacing the current strain, but also adding to existing variant: “Further, excess SGTF
growth rates generally outweighed declines in non-SGTF positives, showing B.1.1.7/VOC202012/01 is
likely adding to, rather than replacing, existing strains”.
(9)
UK variant features
• The B.1.1.7 variant is characterised by a set of 17 mutations present across several genes
(2). Many of
these mutations have been identified before in varying frequencies
(2), but the large number and
1982
combination of these in a variant is new
(8). Several of the genetic changes occur in the spike protein
(4)
(8), which the virus uses to enter host cells.
ACT
• Three key genetic changes to the spike protein include a mutation at position 501, a deletion at
position 69-70, and P681H. The mutation N501Y is in the receptor binding domain of the spike
protein. This may be particularly significant because theoretically, changes in this part of the spike
protein may make the virus more easily transmissible
(7). N501Y has been associated with increased
infectivity and virulence in a mouse model (4). This mutation has also been reported in So
ation Act 1982 uth Africa,
Australia, Denmark, Brazil and the US
(2). H69del/V70del may be associated with immune response
evasion
(9). The spike protein deletion at position 69-70 also affects PCR assays targeting the S-gene
by preventing probe binding and causing S-gene target failure (SGTF)
(1, 9). SGTF
INFORMATION can be used as a
proxy to screen for VOC 202012/01, though whole genome sequencing is more definitive.
Transmissibility of the UK variant
• Preliminary analysis in the UK indicated that this variant is significantly more transmissible than
other variants – it may increase transmissibility up to 30
OFFICIAL -50%, and increase the reproductive number
by 0.4
(10).
(6).
• The proportion of cases tested which have
THE the proxy S gene target failure (SGTF) – a proxy target
that can be reliably used to identify VOC 202012/01 - continued to rise through December in
England. In the first week of December approximately 27.7% of cases contained SGTF, rising to
71.5% in the week 29 December 2020 to 4 Jan
under the O uary 2021
(6).
UNDER
• The ‘secondary attack rate’ (SAR) is the percentage of contacts of a case who become infected. A
secondary attack analysis by Public Health England estimated that 14.7% of the contacts of a case
with VOC 202012/01 become infected, compared to 11% of contacts of a wild type case
(6).
However, the study did not cite whether these were close or causal contacts, and it is unclear what
biases may affect this dat
Released a (e.g. focusing testing on areas where B.1.1.7 is common will result in
higher proportions of tests with that variant).
RELEASED
• Other countries that prioritise genome sequencing, notably Denmark, have also seen a rapid rise in
the proportion of B.1.1.7 variants in their sequencing data, which increases confidence that the
lineage is in fact more transmissible, rather than it becoming more common in the UK due to other
factors.
• A pre-print posted by Volz et al on 4 January examined epidemiological evidence for the lineage
B.1.1.7 having a transmission advantage, through several analyses
(11). All indicated that this VOC
has a substantial transmission advantage. The key metric is that the Ro (reproduction number) of this
link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25 link to page 25
Document 2
VOC was 0.4-0.7 higher than previously circulating variants, which is a significant shift. The ratio of
reproduction numbers varied between 1.4 and 1.8.
Volz et al also note a small but statistically significant shift towards higher rates of infection by the
VOC in those under 20, compared to non VOC. However, it is possible that this is related to
movement/ mixing of this cohort, rather than viral genetics.
• A pre-print posted on 27 December 2020 reported that individuals with B.1.1.7 had higher viral
loads (which may result in more viral shedding from infected individuals, and may translate to higher
infective doses)
(4).
• Another pre-print from 15 January 20201 concluded that “direct population-representative
1982
estimates show that the B.1.1.7/VOC202012/01 SARS-CoV-2 variant leads to higher infection rates,
but does not seem particularly adapted to any age group”; there was no evidence that the rates of
ACT
the new variant were growing faster or slower in those under and over high school age.
(9)
• Some countries have reported increases in the relative frequency of B.1.1.7, but several factors may
affect reporting estimates
(5). O’Toole et al (2021) note that the number of variant genome sequences
reported in each countries will be influenced by the amount of local genomic surveillance, potential
ation Act 1982
targeting of sequencing towards travellers from certain countries, the amount of international travel
among affected countries, and the amount of local transmission
(5)
INFORMATION
Other variants
• On 18 December 2020, the South African government reported the emergence and rapid increase of
another new variant identified through routine genomic surveillance, designated 501.V2
(1). This
OFFICIAL
variant also has multiple changes in the spike protein, including the same N501Y mutation present
in VOC 202012/01. The South African variant (also known as 20H/501Y.V2 or B.1.351) appears to be
THE
more transmissible
(12); One preprint estimates that the South African variant is 50% more
transmissible than previous variants
(13) similar to the increased transmissibility of the UK variant.
• The South African variant contains multiple mutations affecting the spike protein, including K417T,
under the O
E484K, N501Y. One of the mutations, E484K, has the potential to reduce antibody recognition, and it
UNDER
may help SARS-CoV-2 to bypass immune protection provided by prior infection or vaccination.
(14)
As of 7 January 2021, 13 countries had reported B.1.351/501Y.V2
(5).
• In a preliminary study evaluating the effectiveness of the Pfizer/BioNTech vaccine against the N501Y
mutation, found in both the UK and South African variants, researchers found no reduction in
Released
neutralizing activity against the virus with the N501Y mutation
(15). However, this study did not
RELEASED
evaluate the full set of observed spike mutations.
• A Brazilian variant (known as P.1 lineage or 20J/501Y.V3) also has multiple mutations affecting the
spike protein and shares the same K417T, E484K, and N501Y spike protein mutations as the South
African variant. There is evidence to suggest that some of the mutations in the P.1 variant may affect
its transmissibility and antigenic profile, which may affect the ability of antibodies generated
through a previous natural infection or through vaccination to recognize and neutralize the virus
(16).
• A recent study reported on a cluster of cases in Manaus, the largest city in the Amazon region, in
which the P.1 variant was identified in 42% of the specimens sequenced from late December. In this
link to page 25 link to page 25
Document 2
region, it is estimated that approximately 75% of the population had been infected with SARS-CoV2
as of October 2020. However, since mid-December the region has observed a surge in cases. The
emergence of this variant raises concerns of a potential increase in transmissibility or propensity for
SARS-CoV-2 re-infection of individuals
(17).
Implications of new variants
• New variants such as the B.1.1.7 lineage are having significant implications for managing the COVID-
19 pandemic world-wide, for example with increased travel bans internationally, and more elaborate
testing regimens.
1982
• The increased transmissibility would likely lead to greater number of secondary cases and hence
increase the burden on contact tracing; As preliminary data suggests, the number of contacts
infected is 50% higher compared to the current strain (e.g., if the index case has 100 c
ACT ontacts,
number infected increases from, say, 12 previously to 18 with new variant ).
• While there is no evidence that the UK VOC is responsible for more severe disease, the fact that it
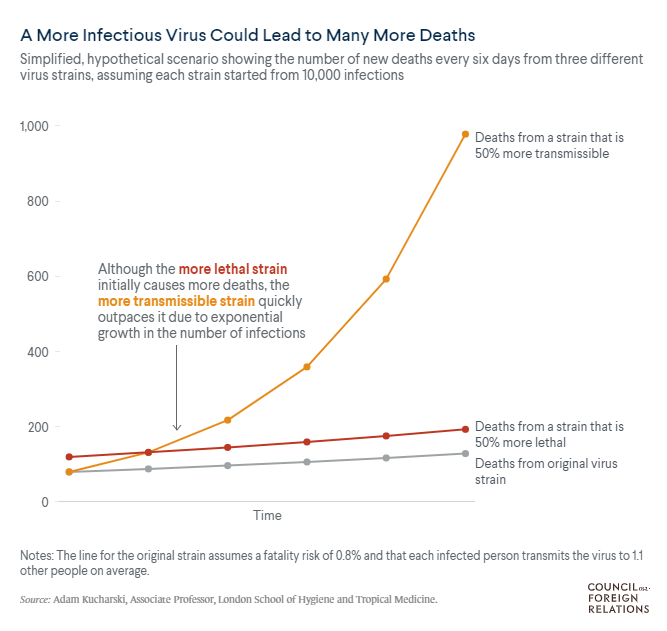
infects more people may result in a higher number of people that need medical care. This is
observed in both the test positivity rates and the hospitalisation numbers out of the UK. A
ation Act 1982 ‘back of
the envelope’ calculation (below) from a mathematical epidemiologist at the London School of
Hygiene and Tropical Medicine, shows that, compared to a virus that infects 10% of contacts with an
0.8% mortality rate, a variant that is 50% more transmissible leads to an increased
INFORMATION number of daily
deaths
(18). Note that this is a very simple analysis for a media brief, comparing hypothetical
exponential growth curves and does not take into account any other factors related to
transmissibility.
OFFICIAL
THE
under the O
UNDER
Released
RELEASED
• The CDC has noted that modelling suggests that lineage B.1.1.7 has the potential to increase the U.S.
pandemic trajectory, warranting universal and increased compliance with mitigation strategies and
suggesting that higher vaccination coverage might be needed to protect the public, i.e., the herd
link to page 25
Document 2
immunity threshold is greater but there is no difference in the individual protection afforded by the
vaccine. The CDC predicts that B.1.1.7 may well become the dominant variant in the US by April-May
2021
(19).
Conclusions
• It seems likely that the UK variant B.1.1.7 has significantly increased transmissibility. The latest evidence
from Public Health England using contact tracing data, estimates that the new variant increases the
number of infected contacts of the index case by 30-50%, and this is seen consistently across
geographic regions in England.
1982
• This coupled with the high rates of infection in the UK, Europe, South Africa and the USA increases
the risk of new cases with this variant arriving in New Zealand.
ACT
• The UK variant is increasingly becoming the dominant variant in the UK, and the US CDC predicts that
it may become the dominant variant there by approximately April 2021. A small number of cases in
New Zealand MIQ have been recorded so far, but this is likely to increase.
ation Act 1982
• New Zealand has imposed pre-departure testing for all long-haul returnees but this will not totally
prevent cases of these variants arriving in New Zealand, although it may reduce the numbers arriving.
• Although there is emerging evidence of increased transmissibility there is no evidence on the need to
INFORMATION
change current quarantine Infection Prevention and Control procedures.
• If there is a breach from MIQ and community transmission occurs, it is likely to spread more rapidly
and be more challenging to contain and also put pressure on contact tracing resources.
• As in other countries, in the setting of an outbreak rapid deployment of a vaccine should be
OFFICIAL
considered to protect vulnerable groups (e.g., community clusters, vulnerable populations such as the
elderly, and Maori and Pasifika communities) and potentially provide some reduction in transmission
rates.
THE
under the O
UNDER
Dr Ian Town Released
Chief Science Advisor
RELEASED
 1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
Document 2
References
1.
European Centre for Disease Prevention and Control. Risk related to spread of new SARSCoV-2 variants of
concern in the EU/EEA - Rapid risk assessment. 2020 29 December 2020. Report No.
2.
World Health Organisation. UK SARS-CoVo2 variant - update from WHO (from the WHO Event Information
Site). 2020 21 December 2020. Report No.
3.
Centers for Disease Control and Prevention. New COVID-19 Variants 2021 [updated 15 January 2021; cited
2021 19 January]. Available from: https://www.cdc.gov/coronavirus/2019-ncov/transmission/variant.html.
4.
Kidd M, Richter A, Best A, Mirza J, Percival B, Mayhew M, et al. S-variant SARS-CoV-2 is associated with
significantly higher viral loads in samples tested by ThermoFisher TaqPath RT-QPCR. medRxiv.
2020:2020.12.24.20248834.
1982
5.
O’Toole A, Hill V, Pybus OG, Watts A, Bogoch II, Khan K, et al. Tracking the international spread of SARS-CoV-
2 lineages B.1.1.7 and B.1.351/501Y-V2. 2021.
6.
Public Health England. Investigation of novel SARS-CoV-2 variant Variant of Concern 202012/01 Technical
ACT
briefing 3. 2021 4 January 2021. Report No.
7.
Wise J. Covid-19: New coronavirus variant is identified in UK. BMJ. 2020;371:m4857.
8.
Rambaut A, Loman N, Pybus O, Barclay W, Barrett J, Carabelli A, et al. Preliminary genomic characterisation
of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. 2020 19 December 2020.
Report No.
ation Act 1982
9.
Walker AS, Vihta K-D, Gethings O, Pritchard E, Jones J, House T, et al. Increased infections, but not viral
burden, with a new SARS-CoV-2 variant. medRxiv. 2021:2021.01.13.21249721.
10.
European Centre for Disease Prevention and Control. Rapid increase of a SARS-CoV-2 variant with multiple
INFORMATION
spike protein mutations observed in the United Kingdom. Stockholm: ECDC, 2020 20 December 2020. Report No.
11.
Volz E, Mishra S, Chand M, Barrett JC, Johnson R, Geidelberg L, et al. Transmission of SARS-CoV-2 Lineage
B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv. 2021:2020.12.30.20249034.
12.
Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, et al. Emergence and rapid spread
of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike
mutations in South Africa. medRxiv. 2020:2020.12.21.20248640.
OFFICIAL
13.
Pearson CAB, Russell, T.W., Davies, N, Kucharski A.J., CMMID COVID-19 working group, Edmunds W.J., Eggo,
R.M. Estimates of severity and transmissibility of novel South Africa SARS-CoV-2 variant 501Y.V2. 2021.
THE
14.
Weisblum Y, Schmidt F, Zhang F, DaSilva J, Poston D, Lorenzi JC, et al. Escape from neutralizing antibodies by
SARS-CoV-2 spike protein variants. eL fe. 2020 Oct 28;9. PubMed PMID: 33112236. Pubmed Central PMCID:
PMC7723407. Epub 2020/10/29. eng.
under the O
15.
Xie X, Zou J, Fontes-Garfias CR, Xia H, Swanson KA, Cutler M, et al. Neutralization of N501Y mutant SARS-
UNDER
CoV-2 by BNT162b2 vaccine elicited sera. bioRxiv. 2021:2021.01.07.425740.
16.
Centers for Disease Control and Prevention. Emerging SARS-CoV-2 Variants 2021 [updated 15 January 2021;
cited 2021 19 January]. Available from: https://www.cdc.gov/coronavirus/2019-ncov/more/science-and-
research/scientific-brief-emerging-variants.html.
17.
Resende PC, Bezerra JF, ., de Vasconcelos RHT, Arantes I, Appolinario L, Mendonça AC, et al. Spike E484K
mutation in the first SARS-CoV-2 reinfection case confirmed in Brazil, 2020 2021 11 January 2021. Available from:
Released
https://virological.org/t/spike-e484k-mutation-in-the-first-sars-cov-2-reinfection-case-confirmed-in-brazil-2020/584.
RELEASED
18.
Felter C. How Dangerous Are New COVID-19 Strains? cfr.org: Council on Foreign Relations; 2021 [cited 2021
20 January 2021]. Available from: https://www.cfr.org/in-brief/how-dangerous-are-new-covid-19-strains.
19.
Galloway SE, Paul P, MacCannell DR, et al. Emergence of SARS-CoV-2 B.1.1.7 Lineage — United States,
December 29, 2020–January 12, 2021. MMWR Morb Mortal Wkly Rep [Internet]. 2021 ePub: 15 January 2021.
Available from:
https://www.cdc.gov/mmwr/volumes/70/wr/mm7003e2.htm?s cid=mm7003e2 e#suggestedcitation
 1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
 1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
 1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
 1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
 1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED

Document 3
The application is being considered for provisional consent under section
23 of the Medicines Act 1981 for the following indications:
Active immunisation to prevent coronavirus disease 2019 (COVID-
19) caused by SARS-CoV-2, in individuals 16 years of age and
older.
The use of the vaccine must be in accordance with official
recommendations.
The application has been submitted via an expedited rolling review
process and has been assessed under urgency due to the significant
clinical need for a COVID 19 vaccine with a positive benefit risk profile.
1982
The initial application was received on 13 November 2020, after which a
total of eight tranches of supporting information were submitted to
Medsafe. Following assessment of these data packages, a request for
ACT
additional information was issued on 15 January 2021 and response from
Pfizer was received on 22 January 2021. Additional responses and data to
support a change in the number of deliverable doses per vial were
received on 27 January 2021. All additional data has since been assessed
and a final recommendation has been made on 28 January 2021.
Given the rapid development of this medicine and the urgent clinical need
that exists in New Zealand, there are several aspects of the data required to
support quality, safety and efficacy that are not available at the time of
INFORMATION
completion of the evaluation. It is also proposed that any provisional
consent be granted for a period of nine months, before which time all
additional data should be received.
It was requested that the Committee focus on the specific aspects in their
consideration of the application:
OFFICIAL
• The conditions and consent time period proposed for provisional
consent under section 23 of the Medicines Act 1981. Specifically,
whether these are appropriate and sufficient given the data
THE
provided up to the time of referral, as well as whether any
additional conditions should be applied.
• Whether the proposed indications for the medicine are appropriate
and supported by the clinical data available, as well as whether any
additional restrictions
UNDER
should be applied.
The following is the full list of evaluation reports and supporting
documentation that were provided:
• Final evaluation report - Quality (includes final recommendation)
•
Final evaluation report - Novel excipients
•
RELEASED Final evaluation report - Non-clinical
• Final evaluation report - Clinical
• Final evaluation report - RMP ·
• Application dossier composed of iterative rolling review tranches
and RFI responses and additional data
• TGA assessment documentation
Medicines Assessment Advisory Committee
Minutes of the 109th meeting on 2 February 2021
Page 4 of 9

Document 3
•
Advice from the Ministry of Health Science and Technical Advisory
Group (STAG) on new SARS-CoV-2 virus strains and the
implications for COVI D-19 vaccines
Pfizer New Zealand was informed of the referral on 29 January 2021.
Medsafe Presentation
Medsafe presented an overview of what is known about Comirnaty to the
Committee.
Pre-clinical discussion
1982
The Committee considered the following documentation:
•
Final evaluation report Non-clinical
ACT
The Committee noted that the pre-clinical questions raised in the report
were addressed satisfactorily by the company. The Committee noted that
pre-clinical observations such as hepatoxicity are not apparent in the clinical
data. The reactogenicity seen in the clinical data does not appear to be a
concern in the pre-clinical data. The data on long terminal half-life of the
lipid nanoparticles was considered unusual but unlikely to be a safety
concern, as only two doses are intended to be administered. The pre-clinical
data did not suggest safety concerns in pregnancy.
INFORMATION
The Committee considers that generally the pre-clinical data has been
superseded by the clinical data. The Committee had no safety concerns
based on the preclinical data.
The Committee adopted the report and agreed with the conclusions.
OFFICIAL
Evaluation
• Quality evaluation report
THE (including Novel Excipients evaluation
report)
The Committee considered the following documentation:
o Final evaluation report Quality (includes final
UNDER
recommendation)
The Committee noted that the Medsafe evaluation report was detailed and
comprehensive. It was noted that many of the questions posed by Medsafe
had been resolved and unresolved questions were included as conditions
for the provisional consent.
The number
RELEASED of quality conditions was noted and that these conditions
addressed instances where usual data was missing due to the developing
nature of the vaccine. The quality conditions align with those required by
other regulators, in particular the European Medicines Agency.
Medsafe noted that under Emergency Use Authorisation procedures,
product released to the US and UK markets are from smaller batch sizes.
Medicines Assessment Advisory Committee
Minutes of the 109th meeting on 2 February 2021
Page 5 of 9

Document 3
The scale difference of the potential New Zealand batches was a focus of
the quality data assessment to ensure vaccine manufactured at commercial
scale is comparable to clinical trial batches.
Pfizer has demonstrated that final product specifications are sufficient to
ensure that product supplied to New Zealand will be comparable to clinical
trial batches. Any gaps in product characterisation would be covered in the
conditions of the provisional consent.
The Committee expressed confidence in the Medsafe quality and
manufacturing evaluation and were interested in being kept informed of
updates in this area.
• Clinical evaluation report
1982
The Committee considered the following documentation:
o Final evaluation report - Clinical
ACT
The Committee considered the issue of efficacy data for subpopulations.
This subset included Maori, Asian, Pacific peoples, the elderly and groups
who are immunocompromised. The Committee commented that the
ethnicity subset data submitted was remarkably similar in efficacy and it is
not unreasonable to assume there is no genetic reason for different
responses in different ethnic groups in New Zealand.
The Committee agreed that it will be important to collect post-market safety
INFORMATION
data for Maori, Pacific peoples, elderly and immunocompromised subsets
as these are the people who are more likely to be at higher risk of
complications of COVID-19. However, the clinical picture on efficacy and
safety will become clearer over time as more people receive the vaccine.
The Committee discussed the lack of data on the duration of response of
OFFICIAL
the vaccine. Medsafe had asked the sponsor for an early cut-off time for
more data, which was not available. The sponsor had confirmed that the
next data analysis from the pivotal clinical trial will arrive in April 2021.
THE
Overall, the Committee was satisfied with the clinical report and summary
presented. The Committee was satisfied with the efficacy data to date
acknowledging that more data will be available over time.
UNDER
• RMP evaluation report
The Committee considered the following documentation:
o Final evaluation report - RMP
The Committee considered that the latest version of the Risk Management
Plan addresses
RELEASED
many areas of concern raised by Medsafe. The need for
additional safety information regarding the elderly, children, people with
comorbidities and immunocompromised people was emphasised.
The Committee noted that patients with autoimmune diseases and patients
who are immunosuppressed were not well represented in clinical trials. The
planned clinical study in patients with rheumatoid arthritis receiving
Medicines Assessment Advisory Committee
Minutes of the 109th meeting on 2 February 2021
Page 6 of9

Document 3
immunomodulators was noted. The Committee expressed concern that
these individuals might be among those prioritised for vaccination before the
results of this study are available. It was noted that this issue is to be
managed as part of the Ministry of Health immunisation implementation
programme.
The need for more information on potential safety signals such as
reactogenicity, anaphylaxis, vaccine-associated enhanced disease and
facial paralysis was noted.
The Committee was satisfied with the updated Risk Management Plan,
noting that additional clinical studies, pharmacovigilance activities and
monthly safety reports are planned to address areas of missing information.
1982
The Committee accepted the Risk Management Plan as written, noting that
it is a living document and there is the opportunity to add safety concerns as
they emerge.
ACT
Discussion with Pfizer
Pfizer representatives joined the meeting to respond to questions from the
Committee. The Committee had questions regarding finished product
testing, risk of transport to New Zealand, in use data in specific populations,
use in severe COVID-19, the emergence of new variants, unforeseen safety
signals after the doses given to date, update on duration of protection and
the new 6 dose proposal. All questions were suitably addressed by Pfizer.
INFORMATION
Discussion to finalise recommendation
Provisional Consent
The Committee unanimously agreed to Medsafe's proposal to grant
OFFICIAL
provisional consent with a nine-month period. This period was proposed to
ensure that all post-approval data commitment deadlines were met. The
Committee agreed with this rationale.
THE
Indications
The Committee agreed that the proposed indication wording for Comirnaty
is revised to the following:
UNDER
Comirnaty has provisional consent (see section 5.1) for the
indication below:
Active immunisation to prevent coronavirus disease 2019 (COV/0-
19) caused by SARS-CoV-2, in individuals 16 years of age and
older.
RELEASED
The use of this vaccine should be in accordance with official
recommendations.
Medicines Assessment Advisory Committee
Minutes of the 109th meeting on 2 February 2021
Page 7 of 9

Document 3
The committee discussed the likely real world use in New Zealand and
acknowledged that the vaccine roll-out will be managed by the Ministry of
Health.
The Committee suggested that Section 5.1 of the data sheet to be revised
to include the following statement:
This medicine has been given a
provisional consent under Section
23 of the Act. This means that further evidence on this medicine is
awaited or that there are specific conditions of use. Refer to the
consent notice published in the New Zealand Gazette for the specific
conditions.
Conditions of provisional consent
1982
The Committee reviewed each proposed condition of the provisional
consent. The Committee agreed that Medsafe could make technical
ACT
amendments to the conditions of consent.
The Committee agreed to the addition of the following condition:
Provide independent batch certification, such as UK National
Institute for Biological Standards and Control (NIBSC) certification,
EU Official Control Authority Batch Release (OCABR) certification,
Australian TGA batch release assessment, or any other certification
agreed with Medsafe, on request for all batches distributed in New
INFORMATION
Zealand.
The Committee raised concerns regarding the wording of the following
conditions.
Provide regular updates on the duration of efficacy and the
OFFICIAL
requirement for booster doses. This should include the six months
analysis data from Study C4591001. Interim report due: April 2021.
THE
Provide updates on efficacy including regarding asymptomatic
infection in the vaccinated group, vaccine failure, immunogenicity,
efficacy in population subgroups and results from post-marketing
studies, when these become available.
UNDER
Submit the final Clinical Study Reports for Study C4591001 and
. Study BNT162-01 once they available.
Inform Medsafe of all safety reviews they conduct or become aware
of and provide the completed review.
The Committee recommended the following amendments to the conditions
RELEASED
to improve clarity of the requirements:
Provide regular reports on the duration of efficacy and the
requirement for booster doses within five working days of these
being produced.
Medicines Assessment Advisory Committee
Minutes of the 109th meeting on 2 February 2021
Page8of9
 1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED

Document 3
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
 1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED

Document 3
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
 1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
 1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED
1982
ACT
INFORMATION
OFFICIAL
THE
UNDER
RELEASED