NEW ZEALAND DATA SHEET
1. PRODUCT NAME
COMIRNATY™ COVID-19 VACCINE 0.5 mg/mL concentrated suspension for injection.
2. QUALITATIVE AND QUANTITATIVE COMPOSITION
This is a multidose vial and must be diluted before use.
One vial (0.45 mL) contains 6 doses of 0.3 mL after dilution, see Section 4.2 Dose and method
of administration and Section 6.6 Special precautions for disposal and other handling.
1 dose (0.3 mL) contains 30 micrograms of BNT162b2 [mRNA] (embedded in lipid
nanoparticles).
The active ingredient is a single-stranded, 5’-capped messenger RNA (mRNA) produced using
a cell-free
in vitro transcription from the corresponding DNA templates, encoding the viral
spike (S) protein of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
For the full list of excipients, see Section 6.1 List of excipients.
3. PHARMACEUTICAL FORM
Concentrated suspension for injection (sterile concentrate).
COMIRNATY is a white to off-white frozen suspension.
4. CLINICAL PARTICULARS
4.1 Therapeutic indications
COMIRNATY has provisional consent (see section 5.1) for the indication below:
Active immunisation to prevent coronavirus disease 2019 (COVID-19) caused by SARS-CoV-
2, in individuals 12 years of age and older.
The use of this vaccine should be in accordance with official recommendations.
4.2 Dose and method of administration
Dose
Individuals 12 years of age and older
COMIRNATY is administered intramuscularly after dilution as a course of 2 doses at least 21
days apart. See dosing instructions below.
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 1 of 19
There are no data available on the interchangeability of COMIRNATY with other COVID-19
vaccines to complete the vaccination course. Individuals who have received 1 dose of
COMIRNATY should receive a second dose of COMIRNATY to complete the vaccination
course.
Elderly population
No dosage adjustment is required in elderly individuals ≥ 65 years of age.
Method of administration
COMIRNATY should be administered intramuscularly after dilution (see Section 6.6 Special
precautions for disposal and other handling).
After dilution, vials of Comirnaty contain six doses of 0.3 mL of vaccine. In order to extract
six doses from a single vial, low dead-volume syringes and/or needles should be used. The low
dead-volume syringe and needle combination should have a dead volume of no more than 35
microlitres. If standard syringes and needles are used, there may not be sufficient volume to
extract a sixth dose from a single vial. Irrespective of the type of syringe and needle:
• Each dose must contain 0.3 mL of vaccine.
• If the amount of vaccine remaining in the vial cannot provide a full dose of 0.3 mL, discard
the vial and any excess volume.
• Do not pool excess vaccine from multiple vials.
The preferred site of administration is the deltoid muscle of the upper arm.
Do not inject COMIRNATY intravascularly, subcutaneously or intradermally.
COMIRNATY should not be mixed in the same syringe with any other vaccines or medicinal
products.
For precautions to be taken before administering COMIRNATY, see Section 4.4 Special
warnings and precautions for use.
For instructions regarding thawing, handling and disposal of COMIRNATY, see Section 6.6
Special precautions for disposal and other handling.
4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of
excipients.
4.4 Special warnings and precautions for use
Traceability
In order to improve the traceability of biological medicinal products, the name and the batch
number of the administered product should be clearly recorded.
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 2 of 19
General recommendations
Hypersensitivity and anaphylaxis
Events of anaphylaxis have been reported. Appropriate medical treatment and supervision
should always be readily available in case of an anaphylactic reaction following the
administration of COMIRNATY.
The individual should be kept under close observation for at least 15 minutes following
vaccination. A second dose of COMIRNATY should not be given to those who have
experienced anaphylaxis to the first dose of COMIRNATY.
Myocarditis and pericarditis
Very rare cases of myocarditis and pericarditis have been observed following vaccination with
COMIRNATY. These cases have primarily occurred within 14 days following vaccination,
more often after the second vaccination, and more often in younger men. Available data suggest
that the course of myocarditis and pericarditis following vaccination is not different from
myocarditis or pericarditis in general.
Healthcare professionals should be alert to the signs and symptoms of myocarditis and
pericarditis. Vaccinees should be instructed to seek immediate medical attention if they develop
symptoms indicative of myocarditis or pericarditis such as (acute and persisting) chest pain,
shortness of breath, or palpitations following vacination. Healthcare professionals should
consult guidance and/or specialists to diagnose and treat this condition.
Stress-related responses
Some individuals may have stress-related responses associated with the process of vaccination
itself. Stress-related responses are temporary and resolve on their own. They may include
dizziness, fainting, palpitations, increases in heart rate, alterations in blood pressure, feeling
short of breath, tingling sensations, sweating and/or anxiety. Individuals should be advised to
bring symptoms to the attention of the vaccination provider for evaluation and precautions
should be in place to avoid injury from fainting.
Concurrent il ness
Vaccination should be postponed in individuals suffering from acute severe febrile illness or
acute infection. The presence of a minor infection and/or low grade fever should not delay
vaccination.
Thrombocytopenia and coagulation disorders
As with other intramuscular injections, COMIRNATY should be given with caution in
individuals receiving anticoagulant ther apy or those with thrombocytopenia or any coagulation
disorder (such as haemophilia) because bleeding or bruising may occur following an
intramuscular administration in these individuals.
Immunocompromised individuals
The efficacy, safety and immunogenicity of COMIRNATY has not been assessed in
immunocompromised individuals, including those receiving immunosuppressant therapy. The
efficacy of COMIRNATY may be lower in immunosuppressed individuals.
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 3 of 19
Duration of protection
The duration of protection afforded by COMIRNATY is unknown as it is still being determined
by ongoing clinical trials.
Limitations of vaccine effectiveness
As with any vaccine, vaccination with COMIRNATY may not protect all vaccine recipients.
Individuals may not be fully protected until 7 days after their second dose of COMIRNATY.
Use in the elderly
Clinical studies of COMIRNATY include participants 65 years of age and older and their data
contributes to the overall assessment of safety and efficacy. See Section 5.1 Pharmacodynamic
properties, Clinical trials, Efficacy against COVID-19. No dosage adjustment is required in
elderly individuals ≥ 65 years of age.
The data for use in the frail elderly (>85 years) is limited. The potential benefits of vaccination
versus the potential risk and clinical impact of even relatively mild systemic adverse events in
the frail elderly should be carefully assessed on a case-by-case basis.
Effects on laboratory tests
No data available.
4.5 Interactions with other medicines and other forms of interactions
No interaction studies have been performed.
Concomitant administration of COMIRNATY with other vaccines has not been studied.
4.6 Fertility, pregnancy and lactation
Fertility
In a combined fertility and developmental toxicity study, female rats were intramuscularly
administered COMIRNATY prior to mating and during gestation (4 full human doses of 30 μg
each, spanning between pre-mating day 21 and gestation day 20). SARS-CoV-2 neutralising
antibodies were present in maternal animals from prior to mating to the end of the study on
postnatal day 21 as well as in fetuses and offspring. There were no vaccine related effects on
female fertility and pregnancy rate.
Pregnancy
There is limited experience with use of COMIRNATY in pregnant women. Animal studies do
not indicate direct or indirect harmful effects with respect to pregnancy, embryo/fetal
development, parturition or post-natal development (see Fertility). Administration of
COMIRNATY in pregnancy should only be considered when the potential benefits outweigh
any potential risks for the mother and fetus.
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 4 of 19
Lactation
It is unknown whether BNT162b2 [mRNA] is excreted in human milk. A combined fertility
and developmental toxicity study in rats did not show harmful effects on offspring development
before weaning (see Fertility).
4.7 Effects on ability to drive and use machines
COMIRNATY has no, or negligible, influence on the ability to drive and use machines.
However, some of the effects mentioned under Section 4.8 Undesirable effects may
temporarily affect the ability to drive or use machines.
4.8 Undesirable effects
Summary of safety profile
The safety of COMIRNATY was evaluated in participants 12 years of age and older in 2
clinical studies that included 22,875 participants (comprised of 21,744 participants 16 years of
age and older and 1,131 adolescents 12 to 15 years of age) that have received at least one dose
of COMIRNATY.
Participants 16 years of age and older
In Study C4591001, a total of 22,026 participants 16 years of age or older received at least 1
dose of COMIRNATY and a total of 22,021 participants 16 years of age or older received
placebo (including 138 and 145 adolescents 16 and 17 years of age in the COMIRNATY and
placebo groups, respectively). A total of 20,519 participants 16 years of age or older received
2 doses of COMIRNATY.
At the time of the analysis of Study C4591001 with a data cut-off of 13 March 2021 for the
placebo-controlled blinded follow-up period up to the participants’ unblinding dates, a total of
25,651 (58.2%) participants (13,031 COMIRNATY and 12,620 placebo) 16 years of age and
older were followed up for ≥4 months after the second dose. This included a total of 15,111
(7,704 COMIRNATY and 7,407 placebo) participants 16 to 55 years of age and a total of
10,540 (5,327 COMIRNATY and 5,213 placebo) participants 56 years and older.
The most frequent adverse reactions in participants 16 years of age and older were injection
site pain (>80%), fatigue (>60%), headache (>50%), myalgia (>40%), chills (>30%), arthralgia
(>20%), pyrexia and injection site swelling (>10%) and were usually mild or moderate in
intensity and resolved within a few days after vaccination. A slightly lower frequency of
reactogenicity events was associated with greater age.
The safety profile in 545 subjects receiving COMIRNATY, that were seropositive for SARS-
CoV-2 at baseline, was similar to that seen in the general population.
Study C4591001 also included 200 participants with confirmed stable human
immunodeficiency virus (HIV) infection. The safety profile of the participants receiving
COMIRNATY (n=100) in the individuals with stable HIV infection was similar to that seen in
the general population.
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 5 of 19
Adolescents 12 through 15 years of age
In an analysis of Study C4591001, 2,260 adolescents (1,131 COMIRNATY; 1,129 placebo)
were 12 through 15 years of age. Of these, 1,308 adolescents (660 COMIRNATY and 648
placebo) have been followed for at least 2 months after the second dose of COMIRNATY. The
safety evaluation in Study C4591001 is ongoing.
The most frequent adverse reactions in adolescents 12 through 15 years of age were injection
site pain (> 90%), fatigue and headache (> 70%), myalgia and chills (> 40%), arthralgia and
pyrexia (> 20%).
Tabulated list of adverse reactions from clinical studies and post-authorisation
experience
Adverse reactions observed during clinical studies are listed below according to the following
frequency categories:
Very common (≥ 1/10),
Common (≥ 1/100 to < 1/10),
Uncommon (≥ 1/1,000 to < 1/100),
Rare (≥ 1/10,000 to < 1/1,000),
Very rare (< 1/10,000),
Not known (cannot be estimated from the available data).
Table 1: Adverse reactions from COMIRNATY clinical trials
Not known
System Organ
Very
Common
Uncommon
Rare
(cannot be
Class
common (≥ 1/100 to
(≥ 1/1,000 to
(≥ 1/10,000 estimated from
(≥ 1/10)
< 1/10)
< 1/100)
to < 1/1,000)
the available
data)
Blood and
Lymphadenopathy
lymphatic
system disorders
Metabolism and
Decreased
nutrition
appetite
disorders
Psychiatric
Insomnia
disorders
Nervous system Headache
Lethargy
Acute
disorders
peripheral
facial
paralysisa
Gastrointestinal
Nausea;
disorders
Skin and
Hyperhidrosis;
subcutaneous
Night sweats
tissue disorders
Musculoskeletal Arthralgia;
and connective
Myalgia
tissue disorders
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 6 of 19
Not known
System Organ
Very
Common
Uncommon
Rare
(cannot be
Class
common (≥ 1/100 to
(≥ 1/1,000 to
(≥ 1/10,000 estimated from
(≥ 1/10)
< 1/10)
< 1/100)
to < 1/1,000)
the available
data)
General
Injection
Injection
Asthenia; Malaise;
disorders and
site pain;
site redness
administration
Fatigue;
site conditions
Chills;
Pyrexiab;
Injection
site
swelling
a Through the clinical trial safety follow-up period to 14 November 2020, acute peripheral facial paralysis (or
palsy) was reported by four participants in the COMIRNATY group. Onset was Day 37 after Dose 1 (participant
did not receive Dose 2) and Days 3, 9, and 48 after Dose 2. No cases of acute peripheral facial paralysis (or palsy)
were reported in the placebo group.
b A higher frequency of pyrexia was observed after the second dose.
Post-marketing experience
Although the events listed in Table 2 were not observed in the clinical trials, they are considered
adverse drug reactions for COMIRNATY as they were reported in the post-marketing
experience. As these reactions were derived from spontaneous reports, the frequencies could
not be determined and are thus considered as not known.
Table 2: Adverse reactions from COMIRNATY post marketing experience
System Organ Class
Adverse Drug Reaction
Immune system disorders
Anaphylaxis
Hypersensitivity reactions (e.g. rash, pruritis, urticaria, angioedema)
Cardiac disorders
Myocarditis
Pericarditis
Gastrointestinal disorders
Diarrhoea
Vomiting
Musculoskeletal and connective Pain in extremity (arm)
tissue disorders
Reporting suspected adverse effects
Reporting suspected adverse reactions after authorisation of the medicine is important. It allows
continued monitoring of the benefit/risk balance of the medicine. Healthcare professionals are
asked to report any suspected adverse reactions at
https://nzphvc.otago.ac.nz/reporting/.
4.9 Overdose
Overdose data is available from 52 study participants included in the clinical trial that due to
an error in dilution received 58 micrograms of COMIRNATY. The COMIRNATY recipients
did not report an increase in reactogenicity or adverse reactions.
In the event of overdose, monitoring of vital functions and possible symptomatic treatment is
recommended.
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 7 of 19
For advice on the management of overdose please contact the National Poisons Centre on 0800
POISON (0800 764766).
5. PHARMACOLOGICAL PROPERTIES
5.1 Pharmacodynamic properties
Pharmacotherapeutic group: vaccines, other viral vaccines, ATC code: J07BX03.
Mechanism of action
The nucleoside-modified messenger RNA in COMIRNATY is formulated in lipid
nanoparticles, which enable delivery of the non-replicating RNA into host cells to direct
transient expression of the SARS-CoV-2 spike (S) antigen. The mRNA codes for membrane-
anchored, full-length S with two point mutations within the central helix. Mutation of these
two amino acids to proline locks S in an antigenically preferred prefusion conformation.
COMIRNATY elicits both neutralising antibody and cellular immune responses to the antigen,
which may contribute to protection against COVID-19.
Clinical efficacy and safety
Efficacy
Study C4591001 is a multicentre, multinational, Phase 1/2/3 randomised, placebo-controlled,
observer-blind dose-finding, vaccine candidate selection and efficacy study in participants 12
years of age and older. Randomisation was stratified by age: 12 through 15 years of age, 16
through 55 years of age, or 56 years of age and older, with a minimum of 40% of participants
in the ≥ 56-year stratum. The study excluded participants who were immunocompromised and
those who had previous clinical or microbiological diagnosis of COVID-19. Participants with
pre-existing stable disease, defined as disease not requiring significant change in therapy or
hospitalisation for worsening disease during the 6 weeks before enrolment, were included as
were participants with known stable infection with human immunodeficiency virus (HIV),
hepatitis C virus (HCV) or hepatitis B virus (HBV).
Efficacy in participants 16 years of age and older
In the Phase 2/3 portion of Study C4591001, based on data accrued through
14 November 2020, approximately 44,000 participants were randomised equally and were to
receive 2 doses of COMIRNATY or placebo. The efficacy analyses included participants that
received their second vaccination within 19 to 42 days after their first vaccination. The majority
(93.1%) of vaccine recipients received the second dose 19 days to 23 days after Dose 1.
Participants are planned to be followed for up to 24 months after Dose 2, for assessments of
safety and efficacy against COVID-19. In the clinical study, participants were required to
observe a minimum interval of 14 days before and after administration of an influenza vaccine
in order to receive either placebo or COMIRNATY. In the clinical study, participants were
required to observe a minimum interval of 60 days before or after receipt of blood/plasma
products or immunoglobulins through to conclusion of the study in order to receive either
placebo or COMIRNATY.
The population for the analysis of the primary efficacy endpoint included, 36,621 participants
12 years of age and older (18,242 in the COMIRNATY group and 18,379 in the placebo group)
who did not have evidence of prior infection with SARS-CoV-2 through 7 days after the second
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 8 of 19
dose. In addition, 134 participants were between the ages of 16 to 17 years of age (66 in the
COMIRNATY group and 68 in the placebo group) and 1616 participants 75 years of age and
older (804 in the COMIRNATY group and 812 in the placebo group).
At the time of the primary efficacy analysis, participants had been followed for symptomatic
COVID19 for in total 2,214 person-years for the COMIRNATY group and in total 2,222
person-years for the placebo group.
There were no meaningful clinical differences in overall vaccine efficacy in participants who
were at risk of severe COVID-19 including those with 1 or more comorbidities that increase
the risk of severe COVID-19 (e.g. asthma, body mass index (BMI) ≥ 30 kg/m2, chronic
pulmonary disease, diabetes mellitus, hypertension).
COMIRNATY efficacy information is presented in Table 3.
Table 3: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2, by age
subgroup – participants without evidence of infection prior to 7 days after Dose 2 –
evaluable efficacy (7 days) population
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Na = 18,198 Cases
Na = 18,325 Cases
Vaccine efficacy
Subgroup
n1b
n1b
%
Surveillance timec
Surveillance timec
(95% CI)f
(n2d)
(n2d)
All participantse
8
162
95.0
2.214 (17,411)
2.222 (17,511)
(90.0, 97.9)
16 to 64 years
7
143
95.1
1.706 (13,549)
1.710 (13,618)
(89.6, 98.1)
65 years and older
1
19
94.7
0.508 (3848)
0.511 (3880)
(66.7, 99.9)
65 to 74 years
1
14
92.9
0.406 (3074)
0.406 (3095)
(53.1, 99.8)
75 years and older
0
5
100.0
0.102 (774)
0.106 (785)
(-13.1, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 [*Case definition: (at least 1 of) fever, new or increased cough,
new or increased shortness of breath, chills, new or increased muscle pain, new loss of taste or smell, sore
throat, diarrhoea or vomiting.]
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the last dose) of
past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not
detected by nucleic acid amplification tests (NAAT) [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of
the surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. No confirmed cases were identified in adolescents 12 to 15 years of age.
f. Two-sided confidence interval (CI) for vaccine efficacy (VE) is derived based on the Clopper and Pearson
method adjusted to the surveillance time. CI not adjusted for multiplicity.
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 9 of 19
In the second primary analysis, efficacy of COMIRNATY in preventing first COVID-19
occurrence from 7 days after Dose 2 compared to placebo was 94.6% (95% credible interval
of 89.9% to 97.3%) in participants 16 years of age and older with or without evidence of prior
infection with SARS-CoV-2.
Additionally, subgroup analyses of the primary efficacy endpoint showed similar efficacy point
estimates across genders, ethnic groups, and participants with medical comorbidities associated
with high risk of severe COVID-19.
Updated efficacy analyses were performed with additional confirmed COVID-19 cases accrued
during blinded placebo-controlled follow-up through 13 March 2021, representing up to
6 months of follow-up after Dose 2 for participants in the efficacy population.
The updated vaccine efficacy information is presented in Table 4.
Table 4: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2, by age
subgroup – participants without evidence of infection and participants with or without
evidence of infection prior to 7 days after Dose 2 – evaluable efficacy (7 days) population
during the placebo-controlled follow-up period
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence
of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Na=20,998
Na=21,096
Cases
Cases
n1b
n1b
Surveil ance Timec
Surveil ance Timec
Vaccine efficacy %
Subgroup
(n2d)
(n2d)
(95% CIe)
All participantsf
77
850
91.3
6.247 (20,712)
6.003 (20,713)
(89.0, 93.2)
16 through 64 years
70
710
90.6
4.859 (15,519)
4.654 (15,515)
(87.9, 92.7)
65 years and older
7
124
94.5
1.233 (4192)
1.202 (4226)
(88.3, 97.8)
65 through 74 years
6
98
94.1
0.994 (3350)
0.966 (3379)
(86.6, 97.9)
75 years and older
1
26
96.2
0.239 (842)
0.237 (847)
(76.9, 99.9)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and
at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or
increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat;
diarrhoea; vomiting).
* Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative
at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = Number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all participants within each group
at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of participants at risk for the endpoint.
e. Two-sided confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method
adjusted to the surveillance time.
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 10 of 19
f. Included confirmed cases in participants 12 through 15 years of age: 0 in the COMIRNATY group (both
without and with or without evidence of prior SARS-CoV-2 infection); 16 and 18 in the placebo group
(without and with or without evidence of prior SARS-CoV-2 infection, respectively).
Efficacy against severe COVID-19
Secondary efficacy analyses suggested benefit of the COVID-19 mRNA Vaccine in preventing
severe COVID-19.
As of 14 November 2020, efficacy against severe COVID-19 (as defined by the study protocol)
occurring after the first dose was 88.9% (95% CI: 20.1, 99.7) (1 case in COVID-19 mRNA
Vaccine group and 9 cases in placebo group), with an estimated vaccine efficacy of 75.0%
(95% CI: -152.6, 99.5) (1 case in COVID-19 mRNA Vaccine group and 4 cases in placebo
group) against severe COVID-19 occurring at least 7 days after Dose 2.
Efficacy against severe COVID-19, defined by the Centers for Disease Control and Prevention
as hospitalisation, admission to the Intensive Care Unit, intubation or mechanical ventilation,
or death occurring after the first dose, was 92.9% (95% CI: 53.2, 99.8) (1 case in COVID-19
mRNA Vaccine group and 14 cases in placebo group).
Efficacy and immunogenicity in adolescents 12 to 15 years of age
An analysis of Study C4591001 has been performed in adolescents 12 to 15 years of age up to
a data cutoff date of 13 March 2021.
The vaccine efficacy information in adolescents 12 to 15 years of age is presented in Table 5.
Table 5: Vaccine efficacy – First COVID-19 occurrence from 7 days after Dose 2 –
participants without evidence of infection and with or without evidence of infection prior
to 7 days after Dose 2 – adolescents 12 to 15 years of age evaluable efficacy (7 days)
population
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
without evidence of prior SARS-CoV-2 infection*
COMIRNATY
Placebo
Na = 1005
Na = 978
Cases
Cases
n1b
n1b
Vaccine efficacy
Surveil ance timec (n2d) Surveil ance timec (n2d)
% (95% CIe)
Adolescents
0
16
12 to 15 years
0.154 (1001)
0.147 (972)
100.0 (75.3, 100.0)
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 to 15 years of age
with or without* evidence of prior SARS-CoV-2 infection
COMIRNATY
Placebo
Na = 1119
Na = 1110
Cases
Cases
n1b
n1b
Vaccine efficacy
Surveil ance timec (n2d) Surveil ance timec (n2d)
% (95% CIe)
Adolescents
0
18
12 to 15 years
0.170 (1109)
0.163 (1094)
100.0 (78.1, 100.0)
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at
least 1 symptom consistent with COVID-19 [*Case definition: (at least 1 of) fever, new or increased cough, new
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 11 of 19
or increased shortness of breath, chills, new or increased muscle pain, new loss of taste or smell, sore throat,
diarrhoea or vomiting).
* Participants who had no serological or virological evidence (prior to 7 days after receipt of the last dose) of
past SARS-CoV-2 infection (i.e, N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not
detected by nucleic acid amplification tests (NAAT) [nasal swab] at Visits 1 and 2), and had negative NAAT
(nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
a. N = number of participants in the specified group.
b. n1 = Number of participants meeting the endpoint definition.
c. Total surveillance time in 1000 person-years for the given endpoint across all subjects within each group at
risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the
surveillance period.
d. n2 = Number of subjects at risk for the endpoint.
e. Confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method adjusted
for surveillance time. CI not adjusted for multiplicity.
In Study C4591001 an analysis of SARS-CoV-2 neutralising titres in a randomly selected
subset of participants was performed to demonstrate non-inferior immune responses (within
1.5-fold) comparing adolescents 12 to 15 years of age to participants 16 to 25 years of age who
had no serological or virological evidence of past SARS-CoV-2 infection. The immune
response to COMIRNATY in adolescents 12 to 15 years of age (n = 190) was non-inferior to
the immune response in participants 16 to 25 years of age (n = 170), based on results for SARS-
CoV-2 neutralising titres at 1 month after Dose 2. The geometric mean titres (GMT) ratio of
the adolescents 12 to 15 years of age group to the participants 16 to 25 years of age group was
1.76, with a 2-sided 95% CI of 1.47 to 2.10, meeting the 1.5-fold non-inferiority criterion (the
lower bound of the 2-sided 95% CI for the geometric mean ratio [GMR] > 0.67), which
indicates a statistically greater response in the adolescents 12 to 15 years of age than that of
participants 16 to 25 years of age.
This medicine has been given a provisional consent under Section 23 of the Act. This means
that further evidence on this medicine is awaited or that there are specific conditions of use.
Refer to the consent notice published in the New Zealand Gazette for the specific conditions.
5.2 Pharmacokinetic properties
Not applicable.
5.3 Preclinical safety data
Genotoxicity/Carcinogenicity
Neither genotoxicity nor carcinogenicity studies were performed. The components of
COMIRNATY (lipids and mRNA) are not expected to have genotoxic potential.
6. PHARMACEUTICAL PARTICULARS
6.1 List of excipients
((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
Distearoylphosphatidylcholine (DSPC)
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 12 of 19
Cholesterol
Potassium chloride
Monobasic potassium phosphate
Sodium chloride
Dibasic sodium phosphate dihydrate
Sucrose
Water for injections
This vaccine contains less than 1 mmol potassium (39 mg) per dose, that is to say essentially
‘potassium-free’.
This vaccine contains less than 1 mmol sodium (23 mg) per dose, that is to say essentially
‘sodium‑free’.
6.2 Incompatibilities
This medicinal product must not be mixed with other medicinal products except those
mentioned in Section 6.6 Special precautions for disposal and other handling.
6.3 Shelf life
Unopened vial
6 months at -90°C to -60°C.
Unopened vials may be stored and transported at -25 C to -15°C for a total of 2 weeks on one
occasion only and can then be returned to -90ºC to -60 C.
Once removed from the freezer, the unopened vial can be stored for up to 1 month at 2°C to
8°C. Within the 1 month shelf-life at 2°C to 8°C, up to 12 hours may be used for transportation.
Prior to use, the unopened vial can be stored for up to 2 hours at temperatures up to 30 C.
Once thawed, COMIRNATY should not be re-frozen.
Diluted medicinal product
Chemical and physical in-use stability, including during transportation, has been demonstrated
for 6 hours at 2ºC to 30 C after dilution in sodium chloride 9 mg/mL (0.9%) solution for
injection. From a microbiological point of view, unless the method of dilution precludes the
risk of microbial contamination, the product should be used immediately. If not used
immediately, in-use storage times and conditions are the responsibility of the user.
6.4 Special precautions for storage
Store in a freezer at -90°C to -60°C.
Store in the original package in order to protect from light.
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 13 of 19
During storage, minimise exposure to room light, and avoid exposure to direct sunlight and
ultraviolet light.
Thawed vials can be handled in room light conditions.
When you are ready to thaw or use COMIRNATY:
Transfers of frozen vials stored at ultra-low temperature (<-60°
C)
• Closed-lid vial trays containing 195 vials removed from ultra-low temperature frozen
storage (<-60°C) may be at temperatures up to 25°C for up to 5 minutes for transfer between
ultra-low-temperature environments.
• Open-lid vial trays, or vial trays containing less than 195 vials removed from ultra-low
temperature frozen storage (<-60°C) may be at temperatures up to <25°C for up to
3 minutes to remove vials or for transfer between ultra-low-temperature environments.
• After vial trays are returned to ultra-low temperature frozen storage following temperature
exposure up to 25°C, they must remain in ultra-low temperature frozen storage for at least
2 hours before they can be removed again.
Transfers of frozen vials stored at -25°C to -15°C
• Closed-lid vial trays containing 195 vials removed from frozen storage (-25°C to -15°C)
may be at temperatures up to 25 C for up to 3 minutes.
• Open-lid vial trays, or vial trays containing less than 195 vials, removed from frozen
storage (-25°C to -15°C) may be at temperatures up to 25°C for up to 1 minute.
Once a vial is removed from the vial tray, it should be thawed for use.
Transportation
If local redistribution of unopened vials is needed, and full trays containing vials cannot be
transported at -90°C to -60°C, available data support physical and chemical stability during
transportation of 1 or more thawed vials at 2°C to 8°C for up to 12 hours. Any hours used for
transport of unopened vials at 2°C to 8°C count against the 1 month limit for storage at 2°C to
8°C.
If local redistribution of diluted medicinal product in vials or syringes is needed, available data
support physical and chemical stability during transportation at 2°C to 30°C for up to 6 hours.
Any hours used for transport of diluted medicinal product in vials or syringes at 2°C to 30°C
count against the 6-hour limit for storage at 2°C and 30°C. Microbiological risks and package
integrity, particularly for prepared dosing syringes, are the responsibility of the preparer during
transportation of diluted medicinal product.
For storage conditions after thawing and dilution of the medicinal product, see Section 6.3
Shelf life.
For additional advice on storing COMIRNATY, contact Pfizer New Zealand on 0800 736 363.
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 14 of 19
 6.5 Nature and contents of container
6.5 Nature and contents of container
2 mL clear vial (Type I glass) with a stopper (synthetic bromobutyl rubber) and a flip-off plastic
cap with aluminium seal. Each vial contains 6 doses, see Section 6.6 Special precautions for
disposal and other handling.
Pack size: 195 vials
6.6 Special precautions for disposal and other handling
COMIRNATY should be prepared by a healthcare professional using aseptic technique to
ensure the sterility of the prepared suspension.
THAWING PRIOR TO DILUTION
• The multidose vial is stored frozen
and must be thawed prior to
No more than
dilution. Frozen vials should be
2 hours at
transferred to an environment of
room
2 °C to 8 °C to thaw; a 195 vial
pack may take 3 hours to thaw.
temperature
Alternatively, frozen vials may also
be thawed for 30 minutes at
(up to 30°C)
temperatures up to 30 °C for
immediate use.
• The unopened vial can be stored for
up to 1 month at 2°C to 8°C.
Within the 1-month shelf-life at 2°C
to 8°C, up to 12 hours may be used
for transportation.
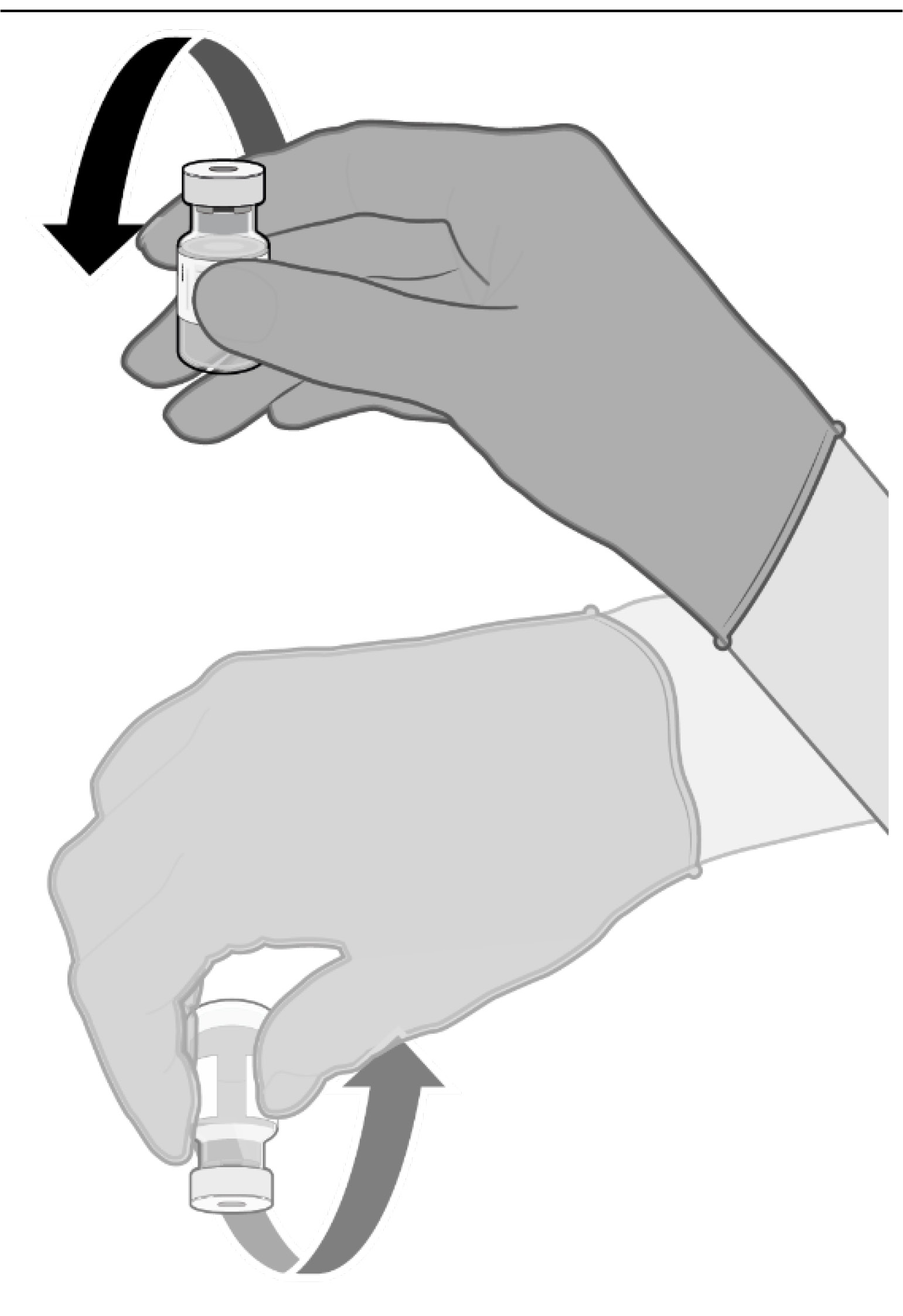
• Allow the thawed vial to come to
room temperature and gently invert
it 10 times prior to dilution.
Do not
shake.
• Prior to dilution, the thawed
suspension may contain white to
off-white opaque amorphous
particles.
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 15 of 19
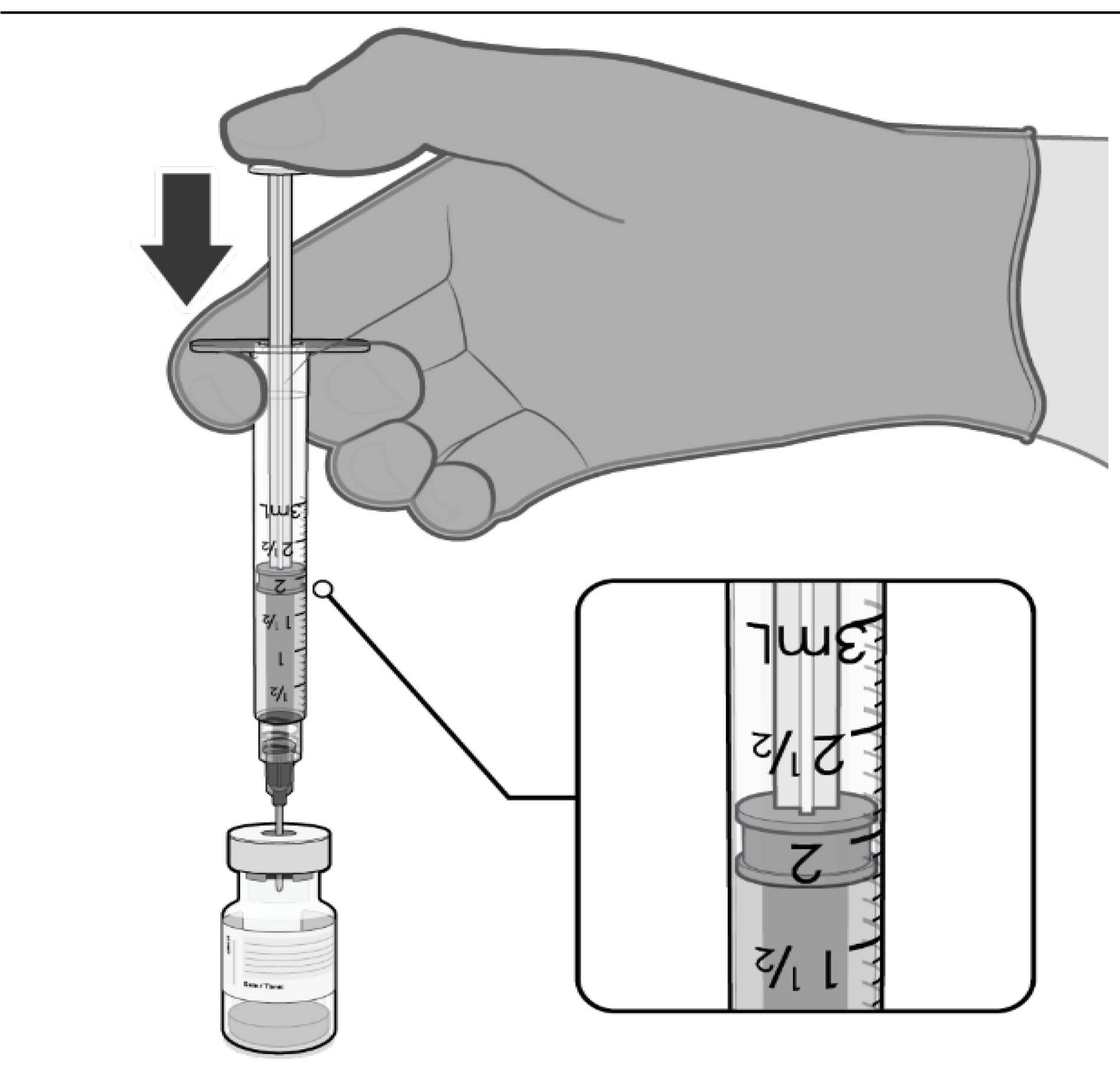

 DILUTION
DILUTION
• The thawed vaccine must be diluted
in its original vial with 1.8 mL
sodium chloride 9 mg/mL (0.9%)
solution for injection, using a
21 gauge or narrower needle and
aseptic techniques. Do not use any
other diluent.
1.8 mL of 0.9% sodium chloride
injection
• Equalise vial pressure before
removing the needle from the vial
stopper by withdrawing 1.8 mL air
into the empty diluent syringe.
Pull back plunger to 1.8 mL to
remove air from vial.
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 16 of 19
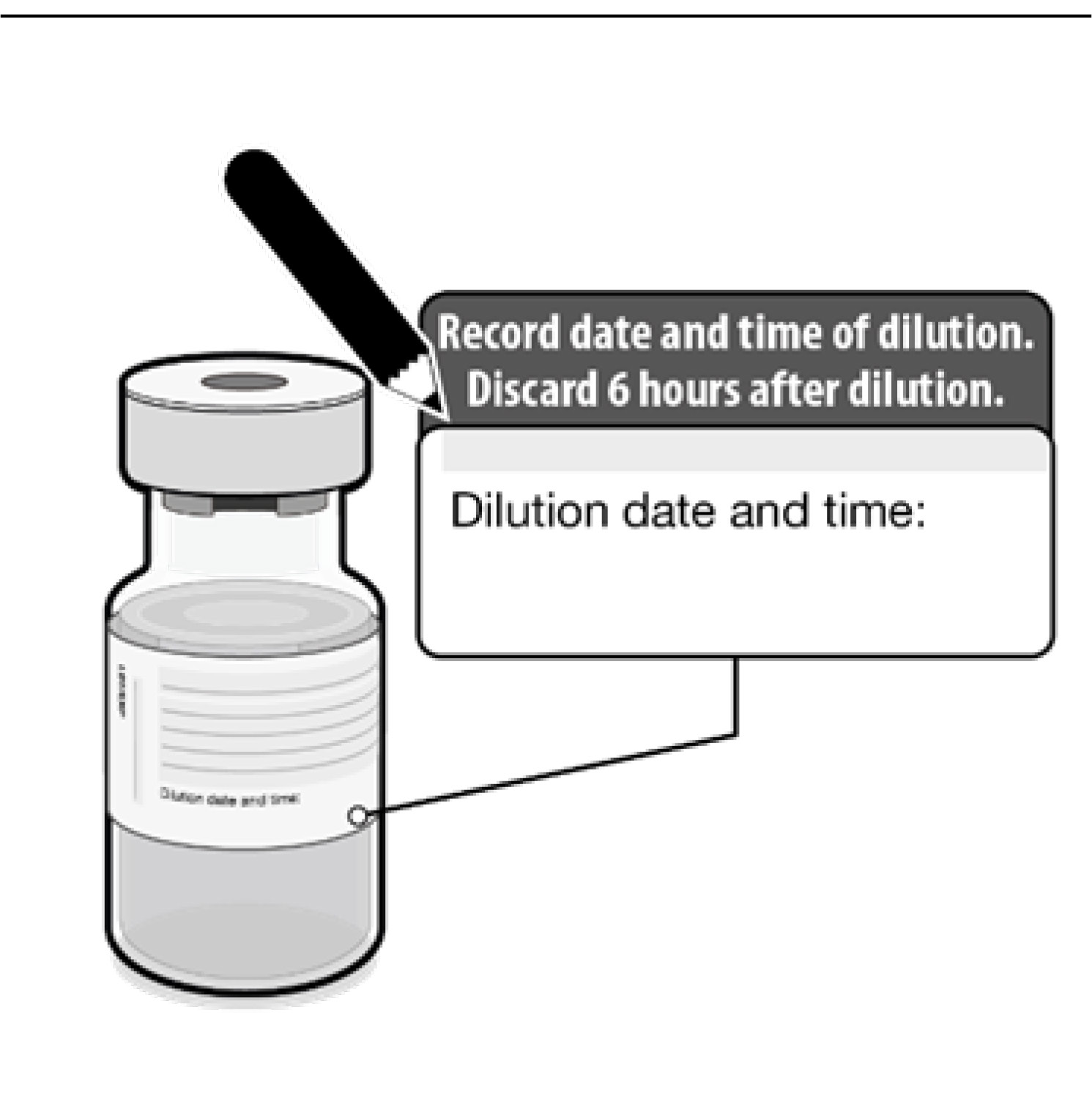
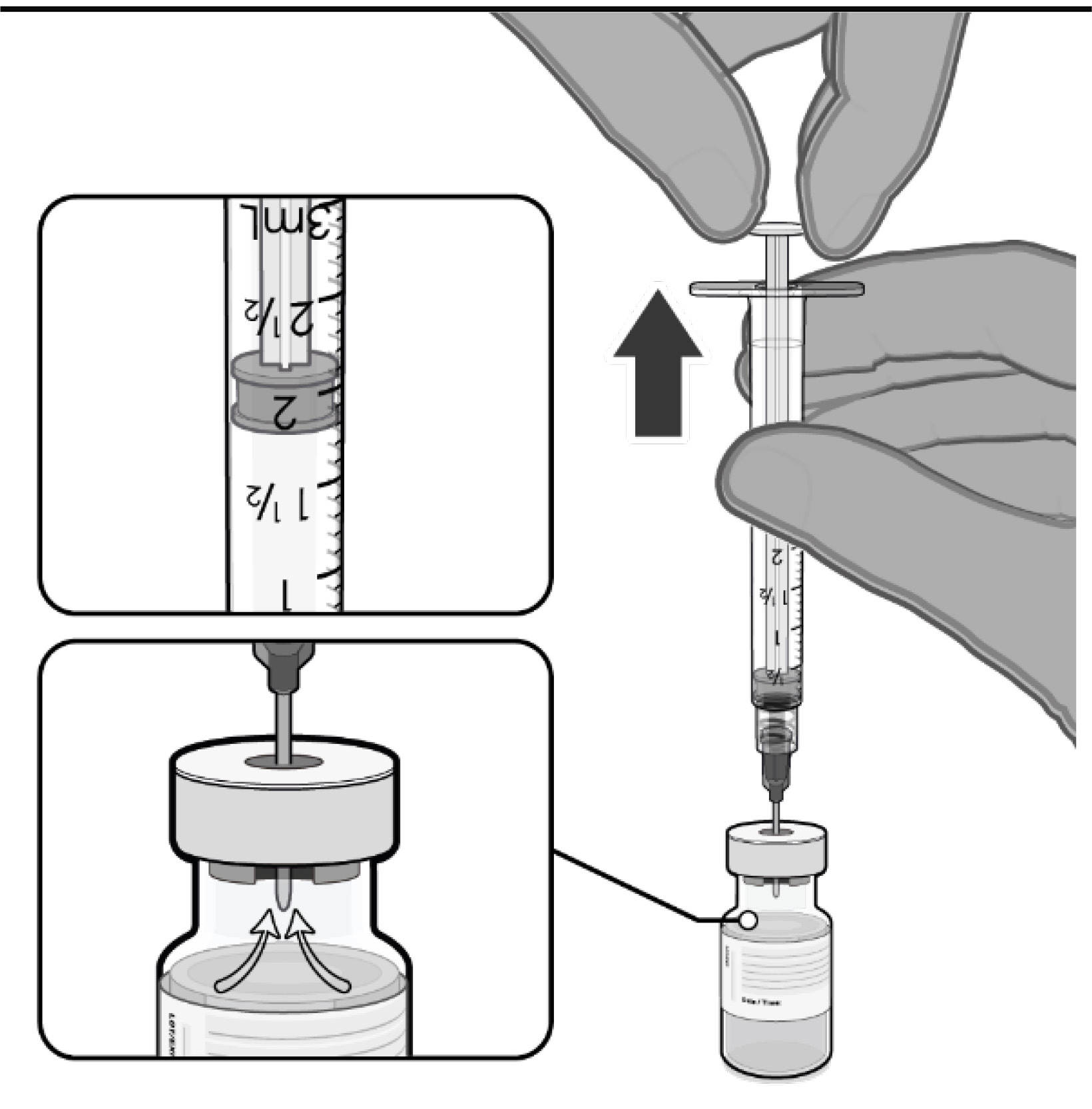
•
Gently invert the diluted
suspension 10 times. Do not shake.
• The diluted vaccine should present
as an off-white suspension with no
particulates visible. Discard the
diluted vaccine if particulates or
discolouration are present.
• The diluted vials should be marked
with the date and time of dilution.
• Do not freeze or shake the diluted
suspension. If refrigerated, allow the
diluted suspension to come to room
temperature prior to use.
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 17 of 19
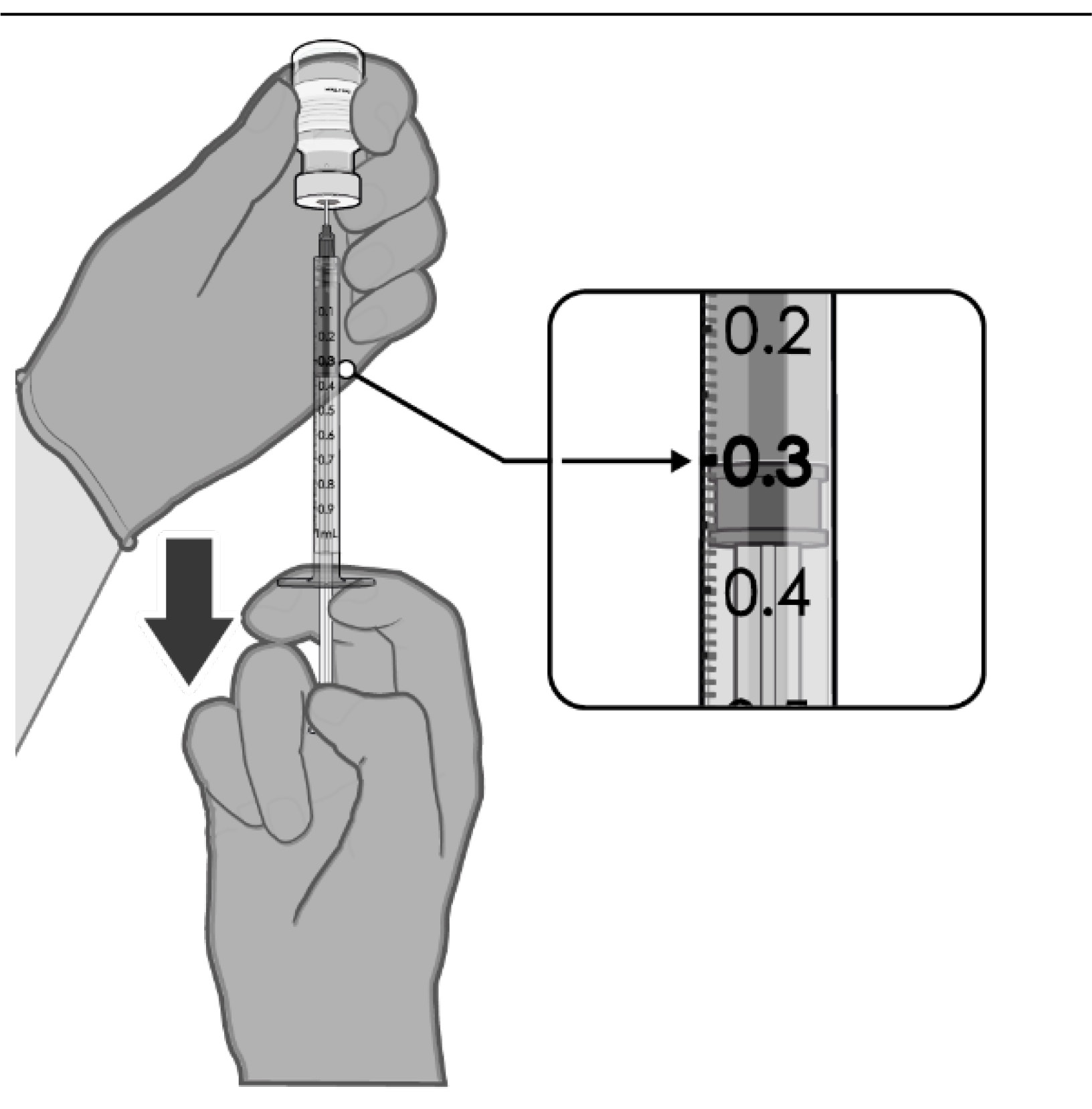 PREPARATION OF INDIVIDUAL 0.3 mL DOSES OF COMIRNATY
PREPARATION OF INDIVIDUAL 0.3 mL DOSES OF COMIRNATY
• After dilution, the vial contains
2.25 mL from which 6 doses of
0.3 mL can be extracted.
• Using aseptic technique, cleanse the
vial stopper with a single-use
antiseptic swab.
• Withdraw 0.3 mL of
COMIRNATY.
Low dead volume syringes and/or
needles should be used in order to
extract 6 doses from a single vial.
The low dead volume syringe and
needle combination should have a
dead volume of no more than 35
microlitres.
If standard syringes and needles are
used, there may not be sufficient
volume to extract a sixth dose from
a single vial.
• Each dose must contain 0.3 mL of
vaccine.
• If the amount of vaccine remaining
in the vial cannot provide a full dose
of 0.3 mL, discard the vial and any
excess volume.
• Verify a final injection volume of
0.3 mL prior to administration.
• Discard syringe and needle after
administration to a single patient.
• Use a new, sterile needle and
syringe to draw up each new dose.
• Discard any unused vaccine 6 hours
after dilution.
Any unused medicine or waste material should be disposed of in accordance with local
requirements.
7. MEDICINE SCHEDULE
Prescription Medicine.
8. SPONSOR
Pfizer New Zealand Limited
P O Box 3998
Auckland, New Zealand
Toll Free Number: 0800 736 363
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 18 of 19
9. DATE OF FIRST APPROVAL
Date of publication in the New Zealand Gazette of consent to distribute this medicine:
03 February 2021
10. DATE OF REVISION OF THE TEXT
28 July 2021
Summary of Updates
Section
Update
4.4
Update for immunisation stress-related responses
Update relating to reports of myocarditis and pericarditis
4.8
Update relating to reports of myocarditis and pericarditis
Updated vaccine safety analyses data in participants followed up to 6
months after Dose 2
5.1
Updated vaccine efficacy analyses data in participants followed up to
6 months after Dose 2
Version: pfdcovii10721
Supersedes: pfdcovii30521
Page 19 of 19